

DNA is often used in solving crimes. But how does DNA profiling actually work?

Professor of Forensic Genetics, Flinders University
Disclosure statement
Adrian Linacre receives funding from the Attorney General's Department of South Australia
Flinders University provides funding as a member of The Conversation AU.
View all partners
DNA profiling is frequently in the news. Public interest is sparked when DNA is used to identify a suspect or human remains , or resolves a cold case that seems all but forgotten.
Very occasionally, it is in the media when the process doesn’t work as it should .
So what is DNA profiling and how does it work – and why does it sometimes not work?
Read more: Australia has 2,000 missing persons and 500 unidentified human remains – a dedicated lab could find matches
A short history of DNA profiling
DNA profiling, as it has been known since 1994, has been used in the criminal justice system since the late 1980s, and was originally termed “DNA fingerprinting”.
The DNA in every human is very similar – up to 99.9% identical , in fact. But strangely, about 98% of the DNA in our cells is not gene-related (i.e. has no known function).
This non-coding DNA is largely comprised of sequences of the four bases that make up the DNA in every cell.

But for reasons unknown, some sections of the sequence are repeated: an example is TCTATCTATCTATCTATCTA where the sequence TCTA is repeated five times. While the number of times this DNA sequence is repeated is constant within a person, it can vary between people. One person might have 5 repeats but another 6, or 7 or 8.
There are a large number of variants and all humans fall into one of them. The detection of these repeats is the bedrock of modern DNA profiling. A DNA profile is a list of numbers, based on the repeated sequences we all have.
The use of these short repeat sequences (the technical term is “ short tandem repeat ” or STR) started in 1994 when the UK Forensic Science Service identified four of these regions . The chance that two people taken at random in the population would share the same repeat numbers at these four regions was about 1 in 50,000.
Now, the number of known repeat sequences has expanded greatly, with the latest test looking at 24 STR regions. Using all of the known STR regions results in an infinitesimally small probability that any two random people have the same DNA profile. And herein lies the power of DNA profiling.

How is DNA profiling performed?
The repeat sequence will be the same in every cell within a person – thus, the DNA profile from a blood sample will be the same as from a plucked hair, inside a tooth, saliva, or skin. It also means a DNA profile will not in itself indicate from what type of tissue it originated.
Consider a knife alleged to be integral to an investigation. A question might be “who held the knife”? A swab (cotton or nylon) will be moistened and rubbed over the handle to collect any cells present.
The swab will then be placed in a tube containing a cocktail of chemicals that purifies the DNA from the rest of the cellular material – this is a highly automated process. The amount of DNA will then be quantified.
If there is sufficient DNA present, we can proceed to generate a DNA profile. The optimum amount of DNA needed to generate the profile is 500 picograms – this is really tiny and represents only 80 cells!

How foolproof is DNA profiling?
DNA profiling is highly sensitive, given it can work from only 80 cells. This is microscopic: the tiniest pinprick of blood holds thousands of blood cells.
Consider said knife – if it had been handled by two people, perhaps including a legitimate owner and a person of interest, yet only 80 cells are present, those 80 cells would not be from only one person but two. Hence there is now a less-than-optimal amount of DNA from either of the people, and the DNA profiling will be a mixture of the two.
Fortunately, there are several types of software to pull apart these mixed DNA profiles. However, the DNA profile might be incomplete (the term for this is “partial”); with less DNA data, there will be a reduced power to identify the person.
Worse still, there may be insufficient DNA to generate any meaningful DNA profile at all. If the sensitivity of the testing is pushed further, we might obtain a DNA profile from even a few cells. But this could implicate a person who may have held the knife innocently weeks prior to an alleged event; or be from someone who shook hands with another person who then held the knife.
This later event is called “indirect transfer” and is something to consider with such small amounts of DNA.
Read more: Criminals can't easily edit their DNA out of forensic databases
What can’t DNA profiling do?
In forensics, using DNA means comparing a profile from a sample to a reference profile, such as taken from a witness, persons of interest, or criminal DNA databases.
By itself, a DNA profile is a set of numbers. The only thing we can figure out is whether the owner of the DNA has a Y-chromosome – that is, their biological sex is male.
A standard STR DNA profile does not indicate anything about the person’s appearance, predisposition to any diseases, and very little about their ancestry.
Other types of DNA testing, such as ones used in genealogy, can be used to associate the DNA at a crime scene to potential genetic relatives of the person – but current standard STR DNA profiling will not link to anyone other that perhaps very close relatives – parents, offspring, or siblings.
DNA profiling has been, and will continue to be, an incredibly powerful forensic test to answer “whose biological material is this”? This is its tremendous strength. As to how and when that material got there, that’s for different methods to sort out.
Read more: New technology lets police link DNA to appearance and ancestry – and it's coming to Australia
- Forensic science
- DNA testing
- DNA profiling
- Criminal justice system
Want to write?
Write an article and join a growing community of more than 183,600 academics and researchers from 4,958 institutions.
Register now
- Introduction to genomics
- In the cell
- Health and disease
- Living things
- Methods and technology
- Science in society
- Genomic conversations
- Resources for 5-12 year olds
- Resources for 13-18 year olds
- Resources for 18+ year olds
- Resources for educators
- Careers in Genomics
- Wellcome Genome Campus
Explore Genomics > Methods and Technology
What is DNA profiling?

DNA profiling is used to identify an individual from a sample of DNA by looking at unique patterns in the DNA sequence.
- On average, about 99.9% of the DNA between two humans is the same. The remaining 0.1% might not sound like much – but that’s around 3 million bases that can differ between two people.
- DNA profiling looks at these differences to produce a pattern that’s unique to an individual. This can be used for solving crime and linking biological relatives.
- Early techniques were called DNA fingerprinting. Just like your actual fingerprint, your DNA fingerprint is something you are born with, and it is unique to you.
What is a DNA fingerprint?
- Each human shares, on average, about 99.9% of their DNA with each other. The remaining 0.1% represents around 3 million bases and is what makes us unique.
- These differences can be compared and used to help distinguish different people from each other.
- One way of doing this is by comparing short sequences of repetitive DNA that show greater variation from one person to the next than other parts of the genome.
- Early techniques produced this pattern by simultaneously comparing sequences of up to 100 bases long, called ‘minisatellites’. This produces a pattern unique to an individual, known as a DNA fingerprint.
- Modern techniques compare even shorter sequences, called ‘microsatellites’.
- Except for identical twins, the probability of two people having the same DNA fingerprint is very small.
- Despite the similar name, a DNA fingerprint has nothing to do with the fingerprint on the tips of the fingers. A DNA fingerprint is the same for every cell in the body and can’t be altered by any known treatment.
How were early DNA fingerprints produced?
- DNA fingerprinting was invented in 1984 by Sir Alec Jeffreys after he realised you could detect variations in human DNA, in the form of minisatellites.
- Early DNA fingerprinting used ‘minisatellites’ – stretches of DNA which are slightly longer than the ‘microsatellites’ used today.
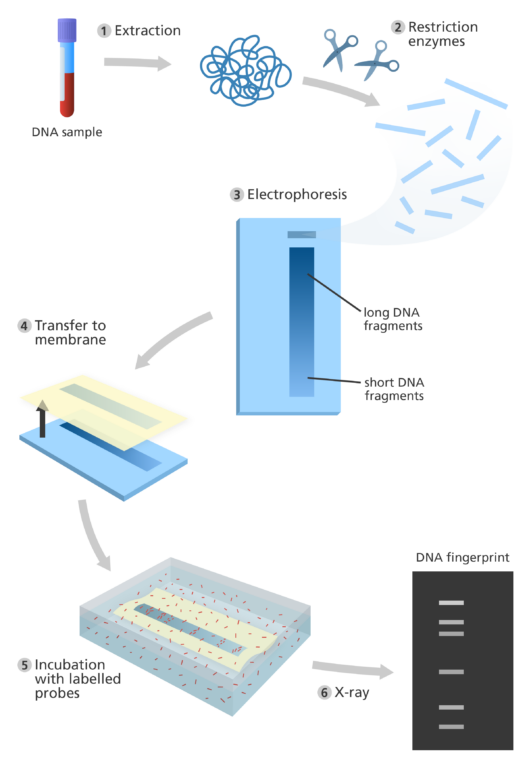
The steps involved in early DNA fingerprinting:
- The first step of DNA fingerprinting was to extract DNA from a sample of human material, usually blood.
- Restriction enzymes – which act like molecular ‘scissors’ – were used to cut the DNA. This resulted in thousands of pieces of DNA with a variety of different lengths.
- The DNA was loaded into wells at one end of a porous gel, which acted a bit like a sieve. An electric current was applied which pulled the negatively charged DNA through the gel.
- The shorter pieces of DNA moved through the gel easiest and fastest. It is more difficult for the longer pieces of DNA to move through the gel, so they travelled slower.
- As a result, by the time the electric current was switched off, the DNA pieces had been separated in order of size. The smallest DNA molecules were furthest away from where the original sample was loaded onto the gel.
- Once the DNA had been separated, the pieces of DNA were transferred from the gel onto a robust piece of nylon. This is called ‘blotting’. The DNA was then ‘unzipped’ to produce single strands of DNA.
- Next the nylon membrane was incubated with radioactive probes – small fragments of microsatellite DNA. They attach to the minisatellite DNA from the genome that they are complementary to.
- The minisatellites that the probes have attached to were then visualised by exposing the nylon membrane to X-ray film. This resulted in a pattern of more than 30 dark bands appearing on the film where the labelled DNA was.
- This pattern was the DNA fingerprint. To compare two or more different DNA fingerprints, the different DNA samples were run side-by-side on the same electrophoresis gel.
Modern DNA profiling
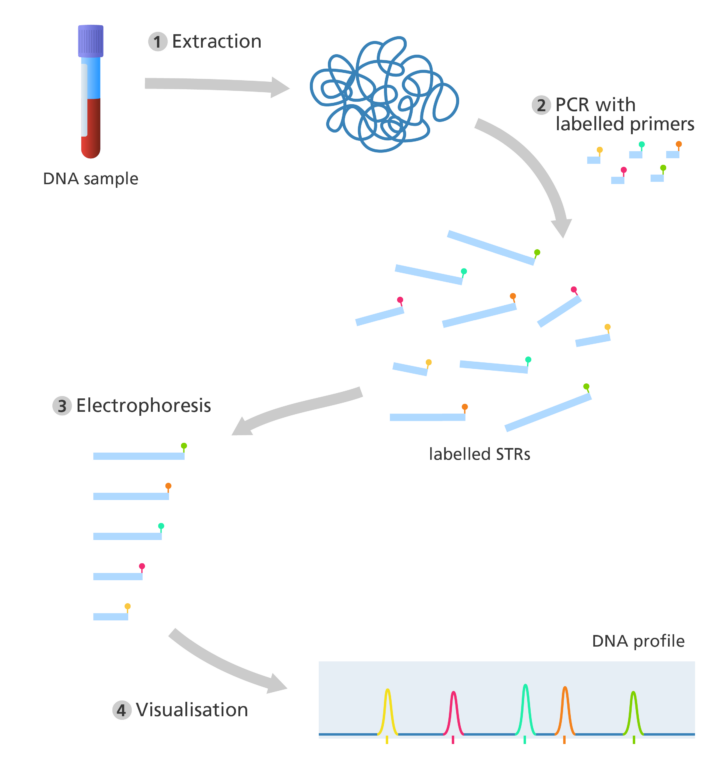
- Modern-day DNA profiling is also called short tandem repeat (STR) analysis.
- It relies on microsatellites, also known as short tandem repeats (STRs). These are the shorter relatives of minisatellites, usually two to five base pairs long.
- Like minisatellites they are repeated many times throughout the human genome, for example ‘TATATATATATA’.
How is a DNA profile produced today?
- DNA is extracted from a biological sample – only a tiny amount of DNA is needed to produce an accurate result. The DNA can be extracted from a wide range of biological samples, including blood, saliva and hair – even if the sample is partially degraded.
- Small pieces of DNA, called ‘primers’, bind to complementary sequences of the DNA either side of the STR of interest. This marks the starting point for the copying of DNA.
- The primers for each STR is labelled with a specific coloured fluorescent tag. This makes it easier to identify and record the STR sequences after PCR.
- Once enough copies of the sequence have been produced, electrophoresis is used to separate the fragments according to size.
- As the DNA fragments separate, they pass by a laser, which causes the fluorescent tags to glow with a specific colour. The output is displayed as a series of coloured peaks (as shown in the image below) highlighting the colour and length of each STR sequence.
How accurate is DNA profiling?
- Only one person in every 10 million million (10,000,000,000,000) will have a particular STR profile.
- It’s extremely unlikely to share the same profile with someone else – except for identical twins.
- About the team
- Copyright information
- Privacy policy
User survey
NOTIFICATIONS
Dna profiling.
- + Create new collection
DNA profiling is the process where a specific DNA pattern, called a profile, is obtained from a person or sample of bodily tissue
Even though we are all unique, most of our DNA is actually identical to other people’s DNA. However, specific regions vary highly between people. These regions are called polymorphic. Differences in these variable regions between people are known as polymorphisms. Each of us inherits a unique combination of polymorphisms from our parents. DNA polymorphisms can be analysed to give a DNA profile.
Human DNA profiles can be used to identify the origin of a DNA sample at a crime scene or test for parentage.
DNA profiling is used to:
- identify the probable origin of a body fluid sample associated with a crime or crime scene
- reveal family relationships
- identify disaster victims, for example, ESR scientists travelled to Thailand to help identify victims of the 2004 Boxing Day tsunami.
What are short tandem repeats?
One of the current techniques for DNA profiling uses polymorphisms called short tandem repeats.
Short tandem repeats (or STRs) are regions of non-coding DNA that contain repeats of the same nucleotide sequence.
For example, GATAGATAGATAGATAGATAGATA is an STR where the nucleotide sequence GATA is repeated six times.
STRs are found at different places or genetic loci in a person’s DNA.
What is a DNA profile?
One way to produce a DNA profile, is for scientists to examine STRs at 10 or more genetic loci. These genetic loci are usually on different chromosomes.
A DNA profile can tell the scientist if the DNA is from a man or woman, and if the sample being tested belongs to a particular person.

Chromosomes
DNA profiling examines sites on several chromosomes.
How do you create a DNA profile using STR?
1. Get a sample of DNA
DNA is found in most cells of the body, including white blood cells, semen, hair roots and body tissue. Traces of DNA can also be detected in body fluids, such as saliva and perspiration because they also contain epithelial cells. Forensic scientists and Police officers collect samples of DNA from crime scenes. DNA can also be collected directly from a person using a mouth swab (which collects inner cheek cells). Find out more in the articles Forensics and DNA and Crime scene evidence .
2. Extract the DNA
DNA is contained within the nucleus of cells. Chemicals are added to break open the cells, extract the DNA and isolate it from other cell components.
3. Copy the DNA
Often only small amounts of DNA are available for forensic analysis so the STRs at each genetic locus are copied many times using the polymerase chain reaction (PCR) to get enough DNA to make a profile. Find out more in the article What is PCR?
Specific primers are used during PCR that attach a fluorescent tag to the copied STRs.
4. Determine the size of the STRs
The size of the STRs at each genetic locus is determined using a genetic analyser. The genetic analyser separates the copied DNA by gel electrophoresis and can detect the fluorescent dye on each STR. This is the same piece of equipment used in the lab for DNA sequencing.
5. Is there a match?
The number of times a nucleotide sequence is repeated in each STR can be calculated from the size of the STRs. A forensic scientist can use this information to determine if a body fluid sample comes from a particular person.
If two DNA profiles from different samples are the same, the chance that the samples came from different people is low. This provides strong evidence that the samples have a common source.

DNA profile
This DNA profile is based on 24 genetic markers (stretches of DNA found at specific locations in the genome). Each genetic marker contains a short tandem repeat (STR)—a section of DNA that repeats itself, like a word typed over and over again, with the number of repeats varying from person to person.
To produce a DNA profile, scientists examine STRs at ten, or more, genetic loci. These genetic loci are usually on different chromosomes.
Activity ideas
Use the activity DNA detective and Mobile forensic kit – unit plan to explore further the use of DNA in solving crimes.
Useful links
ESR is a Crown research institute and is New Zealand’s leading organisation working in forensic science. The information presented above is based on the ESR publication DNA techniques available for use in forensic case work (PDF).
Read about how ESR forensic scientists carry out crime scene investigation .
Two student-friendly Youtube demonstrations on DNA Fingerprinting .
See our newsletters here .
Would you like to take a short survey?
This survey will open in a new tab and you can fill it out after your visit to the site.
DNA Profiling
- Reference work entry
- First Online: 27 November 2018
- pp 1114–1128
- Cite this reference work entry

- Tracy Alexander 5
233 Accesses
1 Citations
DNA identification ; Forensic DNA matching ; Genetic profiling
The use of DNA for identification purposes has often been the subject of controversy. This is particularly true in the UK at present with the imminent implementation of legislation to destroy a significant number of physical samples and to remove the related profiles from the national DNA database. This chapter covers the basic biology behind the use of DNA profiling and its use in the criminal justice system in the UK, particularly in terms of the relevant legislation and the provision of services by private laboratories. These issues are fundamental to the understanding of the problems that have always existed in terms of DNA interpretation, particularly in terms of mixed and partial profiles, plus emerging issues that have arisen with advances in research and technology.
Fundamentals of DNA Profiling in Forensic Science
DNA is a complex chemical compound comprised of relatively simple building blocks...
This is a preview of subscription content, log in via an institution to check access.
Access this chapter
- Available as PDF
- Read on any device
- Instant download
- Own it forever
- Available as EPUB and PDF
- Durable hardcover edition
- Dispatched in 3 to 5 business days
- Free shipping worldwide - see info
Tax calculation will be finalised at checkout
Purchases are for personal use only
Institutional subscriptions
Recommended Reading and References
Butler JM. Fundamentals of forensic DNA typing
Google Scholar
Butler JM, Hill CR (2006) Scientific issues with analysis of low amounts of DNA, 10th edn. American Medical Association
Dror IE (2012) Cognitive forensics and experimental research about bias in forensic casework. Sci Justice 52(2):128–130
Gill P et al (2000) An investigation of the rigor of interpretation rules for STRs derived from less than 100 pg of DNA. Forensic Sci Int 112:17–40
Gross T, Thomson J, Kutranov S (2009) A review of low template STR analysis in casework using the DNA SenCE post-PCR purification technique. Forensic Sci Int: Genet Suppl Ser 2(1):5–7
Jeffreys AJJ (1985a) Hypervariable “minisatellite” regions in human DNA. Nature 314(6006):67–73
Jeffreys AJA (1985b) Positive identification of an immigration test-case using human DNA fingerprints. Nature 317(6040):818–819
Quinones, Daniel (2012) Cell free DNA as a component of forensic evidence recovered from touched surfaces. Forensic Sci Int-Genet 6(1):26–30
Werrett DJ (1997) The national DNA database. Forensic Sci Int 88:33–42
Download references
Author information
Authors and affiliations.
Cold Case Investigation, LGC Forensics, OX14 3ED, Abingdon, UK
Tracy Alexander
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Tracy Alexander .
Editor information
Editors and affiliations.
Netherlands Institute for the Study of Crime and Law Enforcement (NSCR), Amsterdam, The Netherlands
Gerben Bruinsma
VU University Amsterdam, Amsterdam, The Netherlands
Department of Criminology, Law and Society, George Mason University, Fairfax, VA, USA
David Weisburd
Faculty of Law, The Hebrew University, Mt. Scopus, Jerusalem, Israel
Rights and permissions
Reprints and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this entry
Cite this entry.
Alexander, T. (2014). DNA Profiling. In: Bruinsma, G., Weisburd, D. (eds) Encyclopedia of Criminology and Criminal Justice. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5690-2_166
Download citation
DOI : https://doi.org/10.1007/978-1-4614-5690-2_166
Published : 27 November 2018
Publisher Name : Springer, New York, NY
Print ISBN : 978-1-4614-5689-6
Online ISBN : 978-1-4614-5690-2
eBook Packages : Humanities, Social Sciences and Law
Share this entry
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Publish with us
Policies and ethics
- Find a journal
- Track your research
- Australia edition
- Europe edition
- International edition

Eureka moment that led to the discovery of DNA fingerprinting
On 10 September 1984, geneticist Alec Jeffreys wrote three words - "33 autorad off" - in his red desk diary. The phrase marked the completion of an experiment, set up that summer, to study how inherited illnesses pass through families. It failed completely.
Yet the project remains one of the most profoundly influential pieces of research ever carried out in a British laboratory, for it produced the world's first DNA fingerprint, a technology that has revolutionised crime scene investigations, led to the convictions of murderers and rapists, and transformed immigration disputes and paternity cases.
Twenty-five years ago the idea that scientists would one day be able to pinpoint an individual from the tiniest trace of their sweat or blood would have seemed laughable. Today we take it for granted. Along with the CCTV camera and the tapping of emails and phone calls, the DNA fingerprint has become part of a civic apparatus that can follow the movements of individuals with unprecedented accuracy.
Thanks to the research by Jeffreys, thousands of dangerous criminals have been caught and imprisoned and thousands of individuals unfairly denied UK citizenship have been allowed to settle in this country. At the same time, millions of individuals have had their profiles stored in databases in Britain, a serious threat to civil liberties according to some organisations and individuals, a point that is - partially - accepted by Jeffreys himself. More than any other modern scientific discovery, DNA fingerprinting raises crucial issues about balancing the use of technology to help society against an individual's right to privacy.
Such concerns were far from the mind of Alec Jeffreys, then a 34-year-old Leicester University genetics researcher, in the summer of 1984. At the time he was seeking ways to trace genes through family lineages and had hit on a fragment of DNA that was repeated on different chromosomes in the cells of men and women.
This genetic stutter could be unique to an individual, Jeffreys realised, and so he devised an experiment to see if he could count those repeats in different individuals and their relatives, as well as in animals such as seals, mice and monkeys.
First, cells were broken open and their DNA extracted. Then this DNA was attached to photographic films. Radioactive probes - which could identify the repeated sections of DNA - were added. Everything was then placed in a photographic developing tank and left over the weekend of 8-9 September. The results, Jeffreys hoped, would reveal ways that might help him study inherited diseases such as cystic fibrosis.
But when he entered his laboratory that Monday morning and removed the film from its tank, he found an odd array of blobs and lines. "My first reaction was 'God, what a mess.' Then I stared a bit longer - and the penny dropped." That piece of film showed a sequence of bars, each representing different numbers of DNA repeats in the various individuals and animals in the experiment.
Crucially, every individual in the sample had a different bar code and could be identified with precision. Jeffreys could even establish kinships: the bands of the DNA supplied by one of his technicians were a composite of her mother's and father's, for example. Even the animal samples showed that individuals could be identified this way. As Jeffreys put it: "It was an absolute Eureka moment. It was a blinding flash. In five golden minutes, my research career went whizzing off in a completely new direction. The last thing that had been on my mind was anything to do with identification or paternity suits. However, I would have been a complete idiot not to spot the applications."
He called his staff together and they began a brain-storming session to find uses for the technology they had stumbled on. Paternity cases were an obvious example, as was the identification of criminals. "But then we thought, how about crime scene samples. Could we get DNA from blood left behind after murders or robberies?"
Today this seems a silly question, attuned as we are to the marvels displayed in CSI Miami and the rest. But in 1984 no one knew how stable DNA was. For all Jeffreys knew, it could break apart rapidly after a cell had died, making crime scene sampling impossible.
"So I spent the next two days cutting myself and leaving blood marks round the laboratory. Then we tested those bloodstains and found that their DNA was intact." Thus the genetics laboratory of Jeffreys was not only the birthplace of DNA fingerprinting; it became the first setting for a DNA crime scene analysis.
Yet the criminal case uses of DNA fingerprinting were not the first to occupy Jeffreys and his team. Its usefulness in immigration cases grabbed immediate attention. A paper about DNA fingerprinting was written by Jeffreys and his team and was published in Nature in March 1985, triggering several newspaper reports. These were instantly followed up by a group of lawyers who were fighting the deportation of a young boy who, said the Home Office, was not the son of a British woman, as she claimed, and had no right to UK nationality.
"In fact, I had never seen the implications for immigration cases," admits Jeffreys. "It was my wife, Sue, who said DNA fingerprinting would make an incredible difference in disputes over nationality. And she was absolutely right."
After talks with the woman's lawyers, Jeffreys agreed to help. However, the case was complicated by the fact that the boy's father was no longer living in Britain and could not be contacted. "It was like a jigsaw with most of the bits missing," says Jeffreys.
Nevertheless he took samples from the mother, her three daughters and the disputed son. The results "blew me away", he recalls. "It was so incredibly simple. When I looked at the film we made of the DNA samples, I could see that every genetic character in the boy was either present in the woman or in his sisters. He was definitely her son."
The Home Office called in Jeffreys and, after a detailed explanation by him, agreed to drop the case. "Afterwards I went over to tell the mother what had happened, that DNA had done its job," says Jeffreys. "She had a bad time for the past two years and it was clearly affecting her health. But the look on her face when I told her, the relief - it was a magical moment. I realised then that we were on to something of real use. We had reached out and touched someone's life."
Over the next decade, DNA fingerprinting was used to test more than 18,000 immigrants who had been refused entry into the UK. Of these, more than 95% produced results that showed they were blood relatives of UK citizens and were therefore entitled to British citizenship - thanks to DNA fingerprinting.
The next two years were "simply insane", adds Jeffreys. He was inundated with calls from families, mostly of Bangladeshi or Pakistani origins, who had been caught up in immigration disputes. "The university switchboard jammed on several occasions with the calls coming into us." A company, Cellmark, was set up in 1987 to take up these cases and took over much of the Jeffreys caseload.
"Then, out of the blue, I got a call from Leicestershire police, who were investigating the rape and murder of two schoolgirls, Linda Mann and Dawn Ashworth, who lived in the village of Narborough outside Leicester." A local man, Richard Buckland, had just confessed to the murder of Dawn but refused to confess to the killing of Linda. Use your DNA fingerprinting technology to prove he killed both girls, they asked him.
So Jeffreys set up his tests, using - in this case - a version of DNA fingerprinting called DNA profiling. Only a limited number of repeated regions are counted, a technique that is quicker to use and requires smaller samples. It was a perfect opportunity to show off the forensic value of genetic fingerprinting, Jeffreys realised, and, as the tests were being completed, he worked through the night to finish them off as quickly as he could. "I just couldn't wait any longer," he says.
He pulled the film from its developing tank. "I had expected to draw a blank and to find there was not enough DNA in the samples of semen that had been taken from the girls' bodies to produce results." He was wrong. The film was covered in black bands, which showed that the semen from both girls came from the same man, but that Buckland's DNA was completely different. He was not the murderer, the tests indicated. "It was a blood-chilling result," adds Jeffreys.
The police's response was terse and Anglo-Saxon. For his part, the geneticist began to worry that the whole concept of DNA profiling was "up the spout" and that there were things going on biologically that science still did not understand. Then the Home Office repeated the tests and produced the same results as Jeffreys. "I was fretting all the time, but they gave me strength," he adds.
In the end, the police accepted Buckland's innocence and on 21 November 1986, at Leicester Crown Court, he was cleared of the girls' murders. Thus the first use of DNA fingerprinting in a criminal case was to help free an innocent man. "I am pretty sure that, given his confession, Buckland would still be in jail today," adds Jeffreys. "Worse, the real perpetrator would have gone on to kill again."
In the end, that perpetrator was caught by a combination of DNA science and "good old-fashioned coppering", as Jeffreys puts it. In January 1987 police asked all local men between 17 and 34 to submit blood for DNA testing in order to eliminate them from their inquiries. By September, 4,000 had provided samples without success - until a chance remark transformed the investigation.
In a pub one day a local man admitted to his mates he had provided blood on behalf of a friend, Colin Pitchfork. One friend told the police, the man and Pitchfork were arrested and the latter's DNA was shown by Jeffreys to match that of the semen from the two girls' bodies. On 23 January 1988 Pitchfork was sentenced to life for the murders of Linda Mann and Dawn Ashworth. "It was the first time on the planet that a criminal investigation had been tackled and solved at a DNA level," says Jeffreys.
Since then, Jeffreys has used DNA profiling to determine a range of intriguing cases. In 1990 he showed that DNA from bones dug up from a Brazilian graveyard by Nazi-hunters was almost certainly that of Josef Mengele, a doctor who had tortured inmates at Auschwitz. A year later, he helped Home Office scientists prove that bones found in a burial pit in Ekaterinburg, 850 miles east of Moscow, were those of the Russian imperial family who had been killed in 1918 during the Russian civil war.
It is a striking body of work, which earned Jeffreys a knighthood in 1994 and which has taken him far from his academic roots and involved him in a startling range of work. He has no regrets, however: "I love it. DNA fingerprinting came out of the blue and turned me round in five minutes flat. There are certain things in science that are historically inevitable, however. I was just lucky that I got to discover DNA fingerprinting. If I hadn't, someone else would have done it by now. I have no illusions about that."
Forensic History
1984 DNA fingerprints are discovered by Alec Jeffreys. At first, these are used extensively to resolve disputed immigration cases.
1987 The first DNA profile is developed, also by Jeffreys. These use pieces of DNA from only a few selected sites on a person's chromosomes. Repetitions of DNA at these sites are counted, producing a set of numbers that acts as a person's DNA identifier. DNA profiles require smaller forensic samples and are quicker to develop. Crucially, they can also be turned into a sequence of numbers, making it possible for a DNA database to be created.
1995 The UK National Criminal Intelligence DNA Database is established and is used to store the profiles of men and women convicted of crimes in England, Wales and Northern Ireland. A separate database is established in Scotland.
2009 The UK national database is now the largest, per head of population, of any country in the world: almost 10% of the population of England, Wales and Northern Ireland is on the database.
- Forensic science
- The Observer
- DNA database
- UK criminal justice
Most viewed
- Open access
- Published: 18 November 2013
DNA fingerprinting in forensics: past, present, future
- Lutz Roewer 1
Investigative Genetics volume 4 , Article number: 22 ( 2013 ) Cite this article
124k Accesses
82 Citations
59 Altmetric
Metrics details
DNA fingerprinting, one of the great discoveries of the late 20th century, has revolutionized forensic investigations. This review briefly recapitulates 30 years of progress in forensic DNA analysis which helps to convict criminals, exonerate the wrongly accused, and identify victims of crime, disasters, and war. Current standard methods based on short tandem repeats (STRs) as well as lineage markers (Y chromosome, mitochondrial DNA) are covered and applications are illustrated by casework examples. Benefits and risks of expanding forensic DNA databases are discussed and we ask what the future holds for forensic DNA fingerprinting.
The past - a new method that changed the forensic world
'“I’ve found it! I’ve found it”, he shouted, running towards us with a test-tube in his hand. “I have found a re-agent which is precipitated by hemoglobin, and by nothing else”,’ says Sherlock Holmes to Watson in Arthur Conan Doyle’s first novel A study in Scarlet from1886 and later: 'Now we have the Sherlock Holmes’ test, and there will no longer be any difficulty […]. Had this test been invented, there are hundreds of men now walking the earth who would long ago have paid the penalty of their crimes’ [ 1 ].
The Eureka shout shook England again and was heard around the world when roughly 100 years later Alec Jeffreys at the University of Leicester, in UK, found extraordinarily variable and heritable patterns from repetitive DNA analyzed with multi-locus probes. Not being Holmes he refrained to call the method after himself but 'DNA fingerprinting’ [ 2 ]. Under this name his invention opened up a new area of science. The technique proved applicable in many biological disciplines, namely in diversity and conservation studies among species, and in clinical and anthropological studies. But the true political and social dimension of genetic fingerprinting became apparent far beyond academic circles when the first applications in civil and criminal cases were published. Forensic genetic fingerprinting can be defined as the comparison of the DNA in a person’s nucleated cells with that identified in biological matter found at the scene of a crime or with the DNA of another person for the purpose of identification or exclusion. The application of these techniques introduces new factual evidence to criminal investigations and court cases. However, the first case (March 1985) was not strictly a forensic case but one of immigration [ 3 ]. The first application of DNA fingerprinting saved a young boy from deportation and the method thus captured the public’s sympathy. In Alec Jeffreys’ words: 'If our first case had been forensic I believe it would have been challenged and the process may well have been damaged in the courts’ [ 4 ]. The forensic implications of genetic fingerprinting were nevertheless obvious, and improvements of the laboratory process led already in 1987 to the very first application in a forensic case. Two teenage girls had been raped and murdered on different occasions in nearby English villages, one in 1983, and the other in 1986. Semen was obtained from each of the two crime scenes. The case was spectacular because it surprisingly excluded a suspected man, Richard Buckland, and matched another man, Colin Pitchfork, who attempted to evade the DNA dragnet by persuading a friend to give a sample on his behalf. Pitchfork confessed to committing the crimes after he was confronted with the evidence that his DNA profile matched the trace DNA from the two crime scenes. For 2 years the Lister Institute of Leicester where Jeffreys was employed was the only laboratory in the world doing this work. But it was around 1987 when companies such as Cellmark, the academic medico-legal institutions around the world, the national police, law enforcement agencies, and so on started to evaluate, improve upon, and employ the new tool. The years after the discovery of DNA fingerprinting were characterized by a mood of cooperation and interdisciplinary research. None of the many young researchers who has been there will ever forget the DNA fingerprint congresses which were held on five continents, in Bern (1990), in Belo Horizonte (1992), in Hyderabad (1994), in Melbourne (1996), and in Pt. Elizabeth (1999), and then shut down with the good feeling that the job was done. Everyone read the Fingerprint News distributed for free by the University of Cambridge since 1989 (Figure 1 ). This affectionate little periodical published non-stylish short articles directly from the bench without impact factors and resumed networking activities in the different fields of applications. The period in the 1990s was the golden research age of DNA fingerprinting succeeded by two decades of engineering, implementation, and high-throughput application. From the Foreword of Alec Jeffreys in Fingerprint News , Issue 1, January 1989: 'Dear Colleagues, […] I hope that Fingerprint News will cover all aspects of hypervariable DNA and its application, including both multi-locus and single-locus systems, new methods for studying DNA polymorphisms, the population genetics of variable loci and the statistical analysis of fingerprint data, as well as providing useful technical tips for getting good DNA profiles […]. May your bands be variable’ [ 5 ].

Cover of one of the first issues of Fingerprint News from 1990.
Jeffreys’ original technology, now obsolete for forensic use, underwent important developments in terms of the basic methodology, that is, from Southern blot to PCR, from radioactive to fluorescent labels, from slab gels to capillary electrophoresis. As the technique became more sensitive, the handling simple and automated and the statistical treatment straightforward, DNA profiling, as the method was renamed, entered the forensic routine laboratories around the world in storm. But, what counts in the Pitchfork case and what still counts today is the process to get DNA identification results accepted in legal proceedings. Spectacular fallacies, from the historical 1989 case of People vs. Castro in New York [ 6 ] to the case against Knox and Sollecito in Italy (2007–2013) where literally DNA fingerprinting was on trial [ 7 ], disclosed severe insufficiencies in the technical protocols and especially in the DNA evidence interpretation and raised nolens volens doubts on the scientific and evidentiary value of forensic DNA fingerprinting. These cases are rare but frequent enough to remind each new generation of forensic analysts, researchers, or private sector employees that DNA evidence is nowadays an important part of factual evidence and needs thus intense scrutiny for all parts of the DNA analysis and interpretation process.
In the following I will briefly describe the development of DNA fingerprinting to a standardized investigative method for court use which has since 1984 led to the conviction of thousands of criminals and to the exoneration of many wrongfully suspected or convicted individuals [ 8 ]. Genetic fingerprinting per se could of course not reduce the criminal rate in any of the many countries in the world, which employ this method. But DNA profiling adds hard scientific value to the evidence and strengthens thus (principally) the credibility of the legal system.
The technological evolution of forensic DNA profiling
In the classical DNA fingerprinting method radio-labeled DNA probes containing minisatellite [ 9 ] or oligonucleotide sequences [ 10 ] are hybridized to DNA that has been digested with a restriction enzyme, separated by agarose electrophoresis and immobilized on a membrane by Southern blotting or - in the case of the oligonucleotide probes - immobilized directly in the dried gel. The radio-labeled probe hybridizes to a set of minisatellites or oligonucleotide stretches in genomic DNA contained in restriction fragments whose size differ because of variation in the numbers of repeat units. After washing away excess probe the exposure to X-ray film (autoradiography) allows these variable fragments to be visualized, and their profiles compared between individuals. Minisatellite probes, called 33.6 and 33.15, were most widely used in the UK, most parts of Europe and the USA, whereas pentameric (CAC)/(GTG) 5 probes were predominantly applied in Germany. These so-called multilocus probes (MLP) detect sets of 15 to 20 variable fragments per individual ranging from 3.5 to 20 kb in size (Figure 2 ). But the multi-locus profiling method had several limitations despite its successful application to crime and kinship cases until the middle of the 1990s. Running conditions or DNA quality issues render the exact matching between bands often difficult. To overcome this, forensic laboratories adhered to binning approaches [ 11 ], where fixed or floating bins were defined relative to the observed DNA fragment size, and adjusted to the resolving power of the detection system. Second, fragment association within one DNA fingerprint profile is not known, leading to statistical errors due to possible linkage between loci. Third, for obtaining optimal profiles the method required substantial amounts of high molecular weight DNA [ 12 ] and thus excludes the majority of crime-scene samples from the analysis. To overcome some of these limitations, single-locus profiling was developed [ 13 ]. Here a single hypervariable locus is detected by a specific single-locus probe (SLP) using high stringency hybridization. Typically, four SLPs were used in a reprobing approach, yielding eight alleles of four independent loci per individual. This method requires only 10 ng of genomic DNA [ 14 ] and has been validated through extensive experiments and forensic casework, and for many years provided a robust and valuable system for individual identification. Nevertheless, all these different restriction fragment length polymorphism (RFLP)-based methods were still limited by the available quality and quantity of the DNA and also hampered by difficulties to reliably compare genetic profiles from different sources, labs, and techniques. What was needed was a DNA code, which could ideally be generated even from a single nucleated cell and from highly degraded DNA, a code, which could be rapidly generated, numerically encrypted, automatically compared, and easily supported in court. Indeed, starting in the early 1990s DNA fingerprinting methods based on RFLP analysis were gradually supplanted by methods based on PCR because of the improved sensitivity, speed, and genotyping precision [ 15 ]. Microsatellites, in the forensic community usually referred to short tandem repeats (STRs), were found to be ideally suited for forensic applications. STR typing is more sensitive than single-locus RFLP methods, less prone to allelic dropout than VNTR (variable number of tandem repeat) systems [ 16 ], and more discriminating than other PCR-based typing methods, such as HLA-DQA1 [ 17 ]. More than 2,000 publications now detail the technology, hundreds of different population groups have been studied, new technologies as, for example, the miniSTRs [ 18 ] have been developed and standard protocols have been validated in laboratories worldwide (for an overview see [ 19 ]). Forensic DNA profiling is currently performed using a panel of multi-allelic STR markers which are structurally analogous to the original minisatellites but with much shorter repeat tracts and thus easier to amplify and multiplex with PCR. Up to 30 STRs can be detected in a single capillary electrophoresis injection generating for each individual a unique genetic code. Basically there are two sets of STR markers complying with the standards requested by criminal databases around the world: the European standard set of 12 STR markers [ 20 ] and the US CODIS standard of 13 markers [ 21 ]. Due to partial overlap, they form together a standard of 18 STR markers in total. The incorporation of these STR markers into commercial kits has improved the application of these markers for all kinds of DNA evidence with reproducible results from as less than three nucleated cells [ 22 ] and extracted even from severely compromised material. The probability that two individuals will have identical markers at each of 13 different STR loci within their DNA exceeds one out of a billion. If a DNA match occurs between an accused individual and a crime scene stain, the correct courtroom expression would be that the probability of a match if the crime-scene sample came from someone other than the suspect (considering the random, not closely-related man) is at most one in a billion [ 14 ]. The uniqueness of each person’s DNA (with the exception of monozygotic twins) and its simple numerical codification led to the establishment of government-controlled criminal investigation DNA databases in the developed nations around the world, the first in 1995 in the UK [ 23 ]. When a match is made from such a DNA database to link a crime scene sample to an offender who has provided a DNA sample to a database that link is often referred to as a cold hit. A cold hit is of value as an investigative lead for the police agency to a specific suspect. China (approximately 16 million profiles, the United States (approximately 10 million profiles), and the UK (approximately 6 million profiles) maintain the largest DNA database in the world. The percentage of databased persons is on the increase in all countries with a national DNA database, but the proportions are not the same by the far: whereas in the UK about 10% of the population is in the national DNA database, the percentage in Germany and the Netherlands is only about 0.9% and 0.8%, respectively [ 24 ].

Multilocus DNA Fingerprint from a large family probed with the oligonucleotide (GTG) 5 ( Courtesy of Peter Nürnberg, Cologne Center for Genomics, Germany ).
Lineage markers in forensic analysis
Lineage markers have special applications in forensic genetics. Y chromosome analysis is very helpful in cases where there is an excess of DNA from a female victim and only a low proportion from a male perpetrator. Typical examples include sexual assault without ejaculation, sexual assault by a vasectomized male, male DNA under the fingernails of a victim, male 'touch’ DNA on the skin, and the clothing or belongings of a female victim. Mitochondrial DNA (mtDNA) is of importance for the analyses of low level nuclear DNA samples, namely from unidentified (typically skeletonized) remains, hair shafts without roots, or very old specimens where only heavily degraded DNA is available [ 25 ]. The unusual non-recombinant mode of inheritance of Y and mtDNA weakens the statistical weight of a match between individual samples but makes the method efficient for the reconstruction of the paternal or maternal relationship, for example in mass disaster investigations [ 26 ] or in historical reconstructions. A classic case is the identification of two missing children of the Romanov family, the last Russian monarchy. MtDNA analysis combined with additional DNA testing of material from the mass grave near Yekaterinburg gave virtually irrefutable evidence that the two individuals recovered from a second grave nearby are the two missing children of the Romanov family: the Tsarevich Alexei and one of his sisters [ 27 ]. Interestingly, a point heteroplasmy, that is, the presence of two slightly different mtDNA haplotypes within an individual, was found in the mtDNA of the Tsar and his relatives, which was in 1991 a contentious finding (Figure 3 ). In the early 1990s when the bones were first analyzed, a point heteroplasmy was believed to be an extremely rare phenomenon and was not readily explainable. Today, the existence of heteroplasmy is understood to be relatively common and large population databases can be searched for its frequency at certain positions. The mtDNA evidence in the Romanov case was underpinned by Y-STR analysis where a 17-locus haplotype from the remains of Tsar Nicholas II matched exactly to the femur of the putative Tsarevich and also to a living Romanov relative. Other studies demonstrated that very distant family branches can be traced back to common ancestors who lived hundreds of years ago [ 28 ]. Currently forensic Y chromosome typing has gained wide acceptance with the introduction of highly sensitive panels of up to 27 STRs including rapidly mutating markers [ 29 ]. Figure 4 demonstrates the impressive gain of the discriminative power with increasing numbers of Y-STRs. The determination of the match probability between Y-STR or mtDNA profiles via the mostly applied counting method [ 30 ] requires large, representative, and quality-assessed databases of haplotypes sampled in appropriate reference populations, because the multiplication of individual allele frequencies is not valid as for independently inherited autosomal STRs [ 31 ]. Other estimators for the haplotype match probability than the count estimator have been proposed and evaluated using empirical data [ 32 ], however, the biostatistical interpretation remains complicated and controversial and research continues. The largest forensic Y chromosome haplotype database is the YHRD ( http://www.yhrd.org ) hosted at the Institute of Legal Medicine and Forensic Sciences in Berlin, Germany, with about 115,000 haplotypes sampled in 850 populations [ 33 ]. The largest forensic mtDNA database is EMPOP ( http://www.empop.org ) hosted at the Institute of Legal Medicine in Innsbruck, Austria, with about 33,000 haplotypes sampled in 63 countries [ 34 ]. More than 235 institutes have actually submitted data to the YHRD and 105 to EMPOP, a compelling demonstration of the level of networking activities between forensic science institutes around the world. That additional intelligence information is potentially derivable from such large datasets becomes obvious when a target DNA profile is searched against a collection of geographically annotated Y chromosomal or mtDNA profiles. Because linearly inherited markers have a highly non-random geographical distribution the target profile shares characteristic variants with geographical neighbors due to common ancestry [ 35 ]. This link between genetics, genealogy, and geography could provide investigative leads for investigators in non-suspect cases as illustrated in the following case [ 36 ]:

Screenshot of the 16169 C/T heteroplasmy present in Tsar Nicholas II using both forward and reverse sequencing primers ( Courtesy of Michael Coble, National Institute of Standards and Technology, Gaithersburg, USA ).

Correlation between the number of analyzed Y-STRs and the number of different haplotypes detected in a global population sample of 18,863 23-locus haplotypes.

Screenshot from the YHRD depicting the radiation of a 9-locus haplotype belonging to haplogroup J in Southern Europe.
In 2002, a woman was found with a smashed skull and covered in blood but still alive in her Berlin apartment. Her life was saved by intensive medical care. Later she told the police that she had let a man into her apartment, and he had immediately attacked her. The man was subletting the apartment next door. The evidence collected at the scene and in the neighboring apartment included a baseball cap, two towels, and a glass. The evidence was sent to the state police laboratory in Berlin, Germany and was analyzed with conventional autosomal STR profiling. Stains on the baseball cap and on one towel revealed a pattern consistent with that of the tenant, whereas two different male DNA profiles were found on a second bath towel and on the glass. The tenant was eliminated as a suspect because he was absent at the time of the offense, but two unknown men (different in autosomal but identical in Y-STRs) who shared the apartment were suspected. Unfortunately, the apartment had been used by many individuals of both European and African nationalities, so the initial search for the two men became very difficult. The police obtained a court order for Y-STR haplotyping to gain information about the unknown men’s population affiliation. Prerequisites for such biogeographic analyses are large reference databases containing Y-STR haplotypes also typed for ancestry informative single nucleotide markers (SNP) markers from hundreds of different populations. The YHRD proved useful to infer the population origin of the unknown man. The database inquiry indicated a patrilineage of Southern European ancestry, whereas an African descent was unlikely (Figure 5 ). The police were able to track down the tenant in Italy, and with his help, establish the identity of one of the unknown men, who was also Italian. When questioning this man, the police used the information retrieved from Y-STR profiling that he had shared the apartment in Berlin with a paternal relative. This relative was identified as his nephew. Because of the close-knit relationship within the family, this information would probably not have been easily retrieved from the uncle without the prior knowledge. The nephew was suspected of the attempted murder in Berlin. He was later arrested in Italy, where he had committed another violent robbery.
Information on the biogeographic origin of an unknown DNA could also be retrieved from a number of ancestry informative SNPs (AISNPs) on autosomes or insertion/deletion polymorphisms [ 37 , 38 ] but perhaps even better from so-called mini-haplotypes with only <10 SNPs spanning small molecular intervals (<10 kb) with very low recombination among sites [ 39 ]. Each 'minihap’ behaves like a locus with multiple haplotype lineages (alleles) that have evolved from the ancestral human haplotype. All copies of each distinct haplotype are essentially identical by descent. Thus, they fall like Y and mtDNA into the lineage-informative category of genetic markers and are thus useful for connecting an individual to a family or ancestral genetic pool.
Benefits and risks of forensic DNA databases
The steady growth in the size of forensic DNA databases raises issues on the criteria of inclusion and retention and doubts on the efficiency, commensurability, and infringement of privacy of such large personal data collections. In contrast to the past, not only serious but all crimes are subject to DNA analysis generating millions and millions of DNA profiles, many of which are stored and continuously searched in national DNA databases. And as always when big datasets are gathered new mining procedures based on correlation became feasible. For example, 'Familial DNA Database Searching’ is based on near matches between a crime stain and a databased person, which could be a near relative of the true perpetrator [ 40 ]. Again the first successful familial search was conducted in UK in 2004 and led to the conviction of Craig Harman of manslaughter. Craig Harman was convicted because of partial matches from Harman’s brother. The strategy was subsequently applied in some US states but is not conducted at the national level. It was during a dragnet that it first became public knowledge that the German police were also already involved in familial search strategies. In a little town in Northern Germany the police arrested a young man accused of rape because they had analyzed the DNA of his two brothers who had participated in the dragnet. Because of partial matches between crime scene DNA profiles and these brothers they had identified the suspect. In contrast to other countries, the Federal Constitutional Court of Germany decided in December 2012 against the future court use of this kind of evidence.
Civil rights and liberties are crucial for democratic societies and plans to extend forensic DNA databases to whole populations need to be condemned. Alec Jeffreys early on has questioned the way UK police collects DNA profiles, holding not only convicted individuals but also arrestees without conviction, suspects cleared in an investigation, or even innocent people never charged with an offence [ 41 ]. He also criticized that large national databases as the NDNAD of England and Wales are likely skewed socioeconomically. It has been pointed out that most of the matches refer to minor offences; according to GeneWatch in Germany 63% of the database matches provided are related to theft while <3% related to rape and murder. The changes to the UK database came in the 2012’s Protection of Freedoms bill, following a major defeat at the European Court of Human Rights in 2008. As of May 2013 1.1 million profiles (of about 7 million) had been destroyed to remove innocent people’s profiles from the database. In 2005 the incoming government of Portugal proposed a DNA database containing samples from every Portuguese citizen. Following public objections, the government limited the database to criminals. A recent study on the public views on DNA database-related matters showed that a more critical attitude towards wider national databases is correlated with the age and education of the respondents [ 42 ]. A deeper public awareness on the benefits and risks of very large DNA collections need to be built and common ethical and privacy standards for the development and governance of DNA databases need to be adopted where the citizen’s perspectives are taken into consideration.
The future of forensic DNA analysis
The forensic community, as it always has, is facing the question in which direction the DNA Fingerprint technology will be developed. A growing number of colleagues are convinced that DNA sequencing will soon replace methods based on fragment length analysis and there are good arguments for this position. With the emergence of current Next Generation Sequencing (NGS) technologies, the body of forensically useful data can potentially be expanded and analyzed quickly and cost-efficiently. Given the enormous number of potentially informative DNA loci - which of those should be sequenced? In my opinion there are four types of polymorphisms which deserve a place on the analytic device: an array of 20–30 autosomal STRs which complies with the standard sets used in the national and international databases around the world, a highly discriminating set of Y chromosomal markers, individual and signature polymorphisms in the control and coding region of the mitochondrial genome [ 43 ], as well as ancestry and phenotype inference SNPs [ 44 ]. Indeed, a promising NGS approach with the simultaneous analysis of 10 STRs, 386 autosomal ancestry and phenotype informative SNPs, and the complete mtDNA genome has been presented recently [ 45 ] (Figure 6 ). Currently, the rather high error rates are preventing NGS technologies from being used in forensic routine [ 46 ], but it is foreseeable that the technology will be improved in terms of accuracy and reliability. Time is another essential factor in police investigations which will be considerably reduced in future applications of DNA profiling. Commercial instruments capable of producing a database-compatible DNA profile within 2 hours exist [ 47 ] and are currently under validation for law enforcement use. The hands-free 'swab in - profile out’ process consists of automated extraction, amplification, separation, detection, and allele calling without human intervention. In the US the promise of on-site DNA analysis has already altered the way in which DNA could be collected in future. In a recent decision the Supreme court of the United States held that 'when officers make an arrest supported by probable cause to hold for a serious offense and bring the suspect to the station to be detained in custody, taking and analyzing a cheek swab of the arrestee’s DNA is, like fingerprinting and photographing, a legitimate police booking procedure’ (Maryland v. Alonzo Jay King, Jr.). In other words, DNA can be taken from any arrestee, rightly or wrongly arrested, as a part of the normal booking procedure. Twenty-eight states and the federal government now take DNA swabs after arrests with the aim of comparing profiles to the CODIS database, creating links to unsolved cases and to identify the person (Associated Press, 3 June 2013). Driven by the rapid technological progress DNA actually becomes another metric of quick identification. It remains to be seen whether rapid DNA technologies will alter the way in which DNA is collected by police in other countries. In Germany for example the DNA collection is still regulated by the code of the criminal procedure and the use of DNA profiling for identification purposes only is excluded. Because national legislations are basically so different, a worldwide system to interrogate DNA profiles from criminal justice databases seems currently a very distant project.

Schematic overview of Haloplex targeting and NGS analysis of a large number of markers simultaneously. Sequence data are shown for samples from two individuals and the D3S1358 STR marker, the rs1335873 SNP marker, and a part of the HVII region of mtDNA ( Courtesy of Marie Allen, Uppsala University, Sweden ).
At present the forensic DNA technology directly affects the lives of millions people worldwide. The general acceptance of this technique is still high, reports on the DNA identification of victims of the 9/11 terrorist attacks [ 48 ], of natural disasters as the Hurricane Katrina [ 49 ], and of recent wars (for example, in former Yugoslavia [ 50 ]) and dictatorship (for example, in Argentina [ 51 ]) impress the public in the same way as police investigators in white suits securing DNA evidence at a broken door. CSI watchers know, and even professionals believe, that DNA will inevitably solve the case just following the motto Do Not Ask, it’s DNA, stupid! But the affirmative view changes and critical questions are raised. It should not be assumed that the benefits of forensic DNA fingerprinting will necessarily override the social and ethical costs [ 52 ].
This short article leaves many of such questions unanswered. Alfred Nobel used his fortune to institute a prize for work 'in ideal direction’. What would be the ideal direction in which DNA fingerprinting, one of the great discoveries in recent history, should be developed?
Doyle AC: A study in scarlet, Beeton’s Christmas Annual. 1887, London, New York and Melbourne: Ward, Lock & Co
Google Scholar
Jeffreys AJ, Wilson V, Thein SL: Individual-specific “fingerprints” of Human DNA. Nature. 1985, 314: 67-74. 10.1038/314067a0.
Article CAS PubMed Google Scholar
Jeffreys AJ, Brookfield JF, Semeonoff R: Positive identification of an immigration test-case using human DNA fingerprints. Nature. 1985, 317: 818-819. 10.1038/317818a0.
University of Leicester Bulletin Supplement August/. 2004, September
Jeffreys AJ: Foreword. Fingerprint News. 1989, 1: 1-
Lander ES: DNA fingerprinting on trial. Nature. 1989, 339: 501-505. 10.1038/339501a0.
Balding DJ: Evaluation of mixed-source, low-template DNA profiles in forensic science. Proc Natl Acad Sci U S A. 2013, 110: 12241-12246. 10.1073/pnas.1219739110.
Article PubMed Central CAS PubMed Google Scholar
The innocence project. [ http://www.innocenceproject.org ]
Jeffreys AJ, Wilson V, Thein SL: Hypervariable 'minisatellite’ regions in human DNA. Nature. 1985, 314: 67-73. 10.1038/314067a0.
Schäfer R, Zischler H, Birsner U, Becker A, Epplen JT: Optimized oligonucleotide probes for DNA fingerprinting. Electrophoresis. 1988, 9: 369-374. 10.1002/elps.1150090804.
Article PubMed Google Scholar
Budowle B, Giusti AM, Waye JS, Baechtel FS, Fourney RM, Adams DE, Presley LA, Deadman HA, Monson KL: Fixed-bin analysis for statistical evaluation of continuous distributions of allelic data from VNTR loci, for use in forensic comparisons. Am J Hum Genet. 1991, 48: 841-855.
PubMed Central CAS PubMed Google Scholar
Roewer L, Nürnberg P, Fuhrmann E, Rose M, Prokop O, Epplen JT: Stain analysis using oligonucleotide probes specific for simple repetitive DNA sequences. Forensic Sci Int. 1990, 47: 59-70. 10.1016/0379-0738(90)90285-7.
Wong Z, Wilson V, Patel I, Povey S, Jeffreys AJ: Characterization of a panel of highly variable minisatellites cloned from human DNA. Ann Hum Genet. 1987, 51: 269-288. 10.1111/j.1469-1809.1987.tb01062.x.
Jobling MA, Hurles ME, Tyler-Smith C: Chapter 15: Identity and identification. Human Evolutionary Genetics. 2003, Abingdon: Garland Science, 474-497.
Edwards A, Civitello A, Hammond HA, Caskey CT: DNA typing and genetic mapping with trimeric and tetrameric tandem repeats. Am J Hum Genet. 1991, 49: 746-756.
Budowle B, Chakraborty R, Giusti AM, Eisenberg AJ, Allen RC: Analysis of the VNTR locus D1S80 by the PCR followed by high-resolution PAGE. Am J Hum Genet. 1991, 48: 137-144.
Saiki RK, Bugawan TL, Horn GT, Mullis KB, Erlich HA: Analysis of enzymatically amplified beta-globin and HLA-DQ alpha DNA with allele-specific oligonucleotide probes. Nature. 1986, 324: 163-166. 10.1038/324163a0.
Coble MD, Butler JM: Characterization of new miniSTR loci to aid analysis of degraded DNA. J Forensic Sci. 2005, 50: 43-53.
Butler JM: Forensic DNA Typing: Biology, Technology, and Genetics of STR Markers. 2005, New York: Elsevier Academic Press, 2
Gill P, Fereday L, Morling N, Schneider PM: The evolution of DNA databases - Recommendations for new European STR loci. Forensic Sci Int. 2006, 156: 242-244. 10.1016/j.forsciint.2005.05.036.
Budowle B, Moretti TR, Niezgoda SJ, Brown BL: Proceedings of the Second European Symposium on Human Identification. CODIS and PCR-based short tandem repeat loci: law enforcement tools. 1998, Madison, WI: Promega Corporation, 73-88.
Nagy M, Otremba P, Krüger C, Bergner-Greiner S, Anders P, Henske B, Prinz M, Roewer L: Optimization and validation of a fully automated silica-coated magnetic beads purification technology in forensics. Forensic Sci Int. 2005, 152: 13-22. 10.1016/j.forsciint.2005.02.027.
Martin PD, Schmitter H, Schneider PM: A brief history of the formation of DNA databases in forensic science within Europe. Forensic Sci Int. 2001, 119: 225-231. 10.1016/S0379-0738(00)00436-9.
ENFSI survey on DNA Databases in Europe. 2011, [ http://www.enfsi.eu ], December , published 2012-08-18
Roewer L, Parson W: Internet accessible population databases: YHRD and EMPOP. Encyclopedia of Forensic Sciences. Edited by: Siegel JA, Saukko PJ. 2013, Amsterdam: Elsevier B.V, 2
Calacal GC, Delfin FC, Tan MM, Roewer L, Magtanong DL, Lara MC, Rd F, De Ungria MC: Identification of exhumed remains of fire tragedy victims using conventional methods and autosomal/Y-chromosomal short tandem repeat DNA profiling. Am J Forensic Med Pathol. 2005, 26: 285-291. 10.1097/01.paf.0000177338.21951.82.
Coble MD, Loreille OM, Wadhams MJ, Edson SM, Maynard K, Meyer CE, Niederstätter H, Berger C, Berger B, Falsetti AB, Gill P, Parson W, Finelli LN: Mystery solved: the identification of the two missing Romanov children using DNA analysis. PLoS One. 2009, 4: e4838-10.1371/journal.pone.0004838.
Article PubMed Central PubMed Google Scholar
Haas C, Shved N, Rühli FJ, Papageorgopoulou C, Purps J, Geppert M, Willuweit S, Roewer L, Krawczak M: Y-chromosomal analysis identifies the skeletal remains of Swiss national hero Jörg Jenatsch (1596–1639). Forensic Sci Int Genet. 2013, 7: 610-617. 10.1016/j.fsigen.2013.08.006.
Ballantyne KN, Keerl V, Wollstein A, Choi Y, Zuniga SB, Ralf A, Vermeulen M, de Knijff P, Kayser M: A new future of forensic Y-chromosome analysis: rapidly mutating Y-STRs for differentiating male relatives and paternal lineages. Forensic Sci Int Genet. 2012, 6: 208-218. 10.1016/j.fsigen.2011.04.017.
Budowle B, Sinha SK, Lee HS, Chakraborty R: Utility of Y-chromosome short tandem repeat haplotypes in forensic applications. Forensic Sci Rev. 2003, 15: 153-164.
CAS PubMed Google Scholar
Roewer L, Kayser M, de Knijff P, Anslinger K, Betz A, Caglià A, Corach D, Füredi S, Henke L, Hidding M, Kärgel HJ, Lessig R, Nagy M, Pascali VL, Parson W, Rolf B, Schmitt C, Szibor R, Teifel-Greding J, Krawczak M: A new method for the evaluation of matches in non-recombining genomes: application to Y-chromosomal short tandem repeat (STR) haplotypes in European males. Forensic Sci Int. 2000, 114: 31-43. 10.1016/S0379-0738(00)00287-5.
Andersen MM, Caliebe A, Jochens A, Willuweit S, Krawczak M: Estimating trace-suspect match probabilities for singleton Y-STR haplotypes using coalescent theory. Forensic Sci Int Genet. 2013, 7: 264-271. 10.1016/j.fsigen.2012.11.004.
Willuweit S, Roewer L, International Forensic Y Chromosome User Group: Y chromosome haplotype reference database (YHRD): update. Forensic Sci Int Genet. 2007, 1: 83-87. 10.1016/j.fsigen.2007.01.017.
Parson W, Dür A: EMPOP - a forensic mtDNA database. Forensic Sci Int Genet. 2007, 1: 88-92. 10.1016/j.fsigen.2007.01.018.
Roewer L, Croucher PJ, Willuweit S, Lu TT, Kayser M, Lessig R, de Knijff P, Jobling MA, Tyler-Smith C, Krawczak M: Signature of recent historical events in the European Y-chromosomal STR haplotype distribution. Hum Genet. 2005, 116: 279-291. 10.1007/s00439-004-1201-z.
Roewer L: Male DNA Fingerprints say more. Profiles in DNA. 2004, 7: 14-15.
Phillips C, Fondevila M, Lareu MV: A 34-plex autosomal SNP single base extension assay for ancestry investigations. Methods Mol Biol. 2012, 830: 109-126. 10.1007/978-1-61779-461-2_8.
Pereira R, Phillips C, Pinto N, Santos C, dos Santos SE, Amorim A, Carracedo A, Gusmão L: Straightforward inference of ancestry and admixture proportions through ancestry-informative insertion deletion multiplexing. PLoS One. 2012, 7: e29684-10.1371/journal.pone.0029684.
Pakstis AJ, Fang R, Furtado MR, Kidd JR, Kidd KK: Mini-haplotypes as lineage informative SNPs and ancestry inference SNPs. Eur J Hum Genet. 2012, 20: 1148-1154. 10.1038/ejhg.2012.69.
Maguire CN, McCallum LA, Storey C, Whitaker JP: Familial searching: A specialist forensic DNA profiling service utilising the National DNA Database® to identify unknown offenders via their relatives - The UK experience. Forensic Sci Int Genet. 2013, 8: 1-9.
Jeffreys A: Genetic Fingerprinting. Nat Med. 2005, 11: 1035-1039. 10.1038/nm1005-1035.
Machado H, Silva S: Would you accept having your DNA profile inserted in the National Forensic DNA database? Why? Results of a questionnaire applied in Portugal. Forensic Sci Int Genet. 2013, Epub ahead of print
Parson W, Strobl C, Strobl C, Huber G, Zimmermann B, Gomes SM, Souto L, Fendt L, Delport R, Langit R, Wootton S, Lagacé R, Irwin J: Evaluation of next generation mtGenome sequencing using the Ion Torrent Personal Genome Machine (PGM). Forensic Sci Int Genet. 2013, 7: 632-639. 10.1016/j.fsigen.2013.09.007.
Budowle B, van Daal A: Forensically relevant SNP classes. Biotechniques. 2008, 44: 603-608. 610
Allen M, Nilsson M, Havsjö M, Edwinsson L, Granemo J, Bjerke M: Presentation at the 25th Congress of the International Society for Forensic Genetics. Haloplex and MiSeq NGS for simultaneous analysis of 10 STRs, 386 SNPs and the complete mtDNA genome. 2013, Melbourne, 2–7 September 2013
Bandelt HJ, Salas A: Current next generation sequencing technology may not meet forensic standards. Forensic Sci Int Genet. 2012, 6: 143-145. 10.1016/j.fsigen.2011.04.004.
Tan E, Turingan RS, Hogan C, Vasantgadkar S, Palombo L, Schumm JW, Selden RF: Fully integrated, fully automated generation of short tandem repeat profiles. Investigative Genet. 2013, 4: 16-10.1186/2041-2223-4-16.
Article Google Scholar
Biesecker LG, Bailey-Wilson JE, Ballantyne J, Baum H, Bieber FR, Brenner C, Budowle B, Butler JM, Carmody G, Conneally PM, Duceman B, Eisenberg A, Forman L, Kidd KK, Leclair B, Niezgoda S, Parsons TJ, Pugh E, Shaler R, Sherry ST, Sozer A, Walsh A: DNA Identifications after the 9/11 World Trade Center Attack. Science. 2005, 310: 1122-1123. 10.1126/science.1116608.
Dolan SM, Saraiya DS, Donkervoort S, Rogel K, Lieber C, Sozer A: The emerging role of genetics professionals in forensic kinship DNA identification after a mass fatality: lessons learned from Hurricane Katrina volunteers. Genet Med. 2009, 11: 414-417. 10.1097/GIM.0b013e3181a16ccc.
Huffine E, Crews J, Kennedy B, Bomberger K, Zinbo A: Mass identification of persons missing from the break-up of the former Yugoslavia: structure, function, and role of the International Commission on Missing Persons. Croat Med J. 2001, 42: 271-275.
Corach D, Sala A, Penacino G, Iannucci N, Bernardi P, Doretti M, Fondebrider L, Ginarte A, Inchaurregui A, Somigliana C, Turner S, Hagelberg E: Additional approaches to DNA typing of skeletal remains: the search for “missing” persons killed during the last dictatorship in Argentina. Electrophoresis. 1997, 18: 1608-1612. 10.1002/elps.1150180921.
Levitt M: Forensic databases: benefits and ethical and social costs. Br Med Bull. 2007, 83: 235-248. 10.1093/bmb/ldm026.
Download references
Author information
Authors and affiliations.
Department of Forensic Genetics, Institute of Legal Medicine and Forensic Sciences,, Charité - Universitätsmedizin Berlin,, Berlin,, Germany
Lutz Roewer
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Lutz Roewer .
Additional information
Competing interests.
The author declares that he has no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Authors’ original file for figure 1
Authors’ original file for figure 2, authors’ original file for figure 3, authors’ original file for figure 4, authors’ original file for figure 5, authors’ original file for figure 6, rights and permissions.
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License ( http://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated.
Reprints and permissions
About this article
Cite this article.
Roewer, L. DNA fingerprinting in forensics: past, present, future. Investig Genet 4 , 22 (2013). https://doi.org/10.1186/2041-2223-4-22
Download citation
Received : 08 October 2013
Accepted : 08 October 2013
Published : 18 November 2013
DOI : https://doi.org/10.1186/2041-2223-4-22
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- DNA fingerprinting
- Forensic DNA profiling
- Short tandem repeat
- Lineage markers
- Ancestry informative markers
- Forensic DNA database
- Privacy rights
Investigative Genetics
ISSN: 2041-2223
- Submission enquiries: [email protected]
- General enquiries: [email protected]
Advertisement
How DNA Profiling Works
- Share Content on Facebook
- Share Content on LinkedIn
- Share Content on Flipboard
- Share Content on Reddit
- Share Content via Email

The term "DNA," once used only by scientists, has become part of our everyday lexicon. It's almost impossible to not know of DNA profiling , from the court system to genealogy.
It's also nearly impossible to be unaware of the controversy. Now that we can each have a profile that identifies us solely by our DNA, many people worry about who has access to it and how they might use it.
You probably have a good idea of what DNA is. However, you may not know what type of information DNA evidence yields and how we process and analyze it.
What Is DNA Profiling?
Creating a dna profile, use of dna profiles in law enforcement, use of dna profiles in genealogy, controversy in dna profiling.

Sometimes called DNA fingerprinting or genetic fingerprinting, DNA profiling is simply the collection, processing and analysis of VNTRs — unique sequences on the loci (area on a chromosome).
VNTR stands for variable number tandem repeats — meaning the tandem repeats, or pairs of nucleotides, vary in number.
Most human DNA sequences in different people look too similar to tell apart. After processing, however, VNTRs result in unique bands that make identification possible. In 1984, while looking at the results of an experiment to analyze DNA belonging to different people, Dr. Alec Jeffreys discovered these differences.
"It was an absolute Eureka moment," Jeffreys said, according to The Guardian . "It was a blinding flash. In five golden minutes, my research career went whizzing off in a completely new direction. The last thing that had been on my mind was anything to do with identification or paternity suits. However, I would have been a complete idiot not to spot the applications."
Until the late 1980s — when the commercialization of the technique began — Jeffreys' lab was the only one in the world doing DNA fingerprinting (the original name for DNA profiling, which is not as frequently in use to avoid confusion with actual fingerprinting ).
Although this sounds simple enough, there are several different techniques for creating a DNA profile, and new technology is always emerging.
If they all arrive at a similar result — a unique DNA profile — then why are there so many different techniques for analysis? The best technique for a given application depends on a number of factors, including cost, time available for analysis and the quality and amount of the DNA sample available.
Restriction Fragment Length Polymorphism (RFLP)
The first method for creating a DNA profile was RFLP, or restriction fragment length polymorphism. RFLP is not as common today because it requires a large sample of DNA — as much as 25 hairs or a nickel-sized spot of bodily fluid — and can take as long as a month to complete [source: Baden].
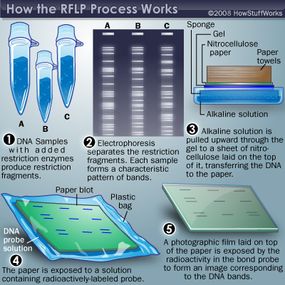
It also requires examining multiple sections of the DNA strand to find variations, which is time-consuming and leaves more room for human error. Some of the steps for RFLP analysis are still in use in other types of DNA profiling.
For RFLP, the steps are:
- Separate white and red blood cells with a centrifuge.
- Extract DNA nuclei from the white blood cells by bathing the cells in hot water, then adding salt, and putting the mixture back into the centrifuge [source: University of Arizona].
- Cut the DNA strand into fragments using a restriction enzyme.
- Place the DNA fragments into one end of a bed of agarose gel with electrodes in it. Agarose gel derives from agar-agar, a type of seaweed that turns into gelatin when dissolved in boiling water.
- Use an electric current to sort the DNA segments by length, a process called agarose gel electrophoresis. Electrophoresis refers to the process of moving the negatively charged molecules through the gel with electricity. Shorter segments move farther away from their original location, while longer ones stay closer. The segments align in parallel rows.
- Use a sheet of nitrocellulose or nylon to blot the DNA. The sheet has stains so that you can see the different lengths of DNA bands with the naked eye. Treating the sheet with radiation creates an autoradiograph, an image on X-ray film left by the decay pattern of the radiation. The autoradiograph, with its distinctive dark-colored parallel bands, is the DNA profile.
Polymerase Chain Reaction (PCR)
PCR analysis is usually the first step in the creation of a DNA profile today. PCR enables DNA amplification, meaning it can replicate a small amount of DNA to create a larger sample for analysis. It does this using a repeating process that takes about five minutes.
- First, you add a heat-stable DNA polymerase — a special enzyme that binds to the DNA and allows it to replicate.
- Next, heat the DNA sample to 200 degrees F (93 degrees C) to separate the threads.
- Then let the cool before reheating it. Reheating doubles the number of copies.
- After repeating this process about 30 times, there is enough DNA for further analysis.
PCR is also the first step in analyzing STRs (Short Tandem Repeats), which are very small, specific alleles in a variable number tandem repeat (VNTR). Alleles are pairs of genes that occur alternately at a specific point, or loci, on a chromosome.
Analyzing STRs is more accurate than the RFLP technique because their small size makes them easier to separate and differentiate.
A variation on STR analysis is Y-STR. Only STRs found on the Y-chromosome (which only males have) is analyzed. STR analysis is useful if the biological samples have mixed DNA (from both men and women) or in sexual assault cases with a male assailant. Y-STR is otherwise processed just like a regular STR.
Amplified Fragment Length Polymorphism (AmpFLP)
AmpFLP, amplified fragment length polymorphism, is another technique that uses PCR to replicate DNA. Like RFLP, it first uses a restriction enzyme. Then, it amplifies the fragments using PCR and sorts them using gel electrophoresis.
AmpFLP's advantage over other techniques is that you can automate the process, and it doesn't cost very much. However, the DNA sample must be high quality or errors may result, which is the case with most DNA analysis techniques.
Analysts can have a hard time telling the longer strands apart because they bunch up tightly.
DNA is often in the news, but one of the most recent stories included a new term: touch DNA. Although it's new to the media, touch DNA has been in existence for several years. Scientists conduct DNA extraction from bodily fluids such as blood and semen, often located by the stains they leave behind.
Touch DNA involves recovering DNA from skin cells left by the perpetrator.
In the JonBenet Ramsey case, investigators scraped clothing that JonBenet had been wearing. There was enough evidence in two different places to create a DNA profile that matched one already created from blood — both of which belonged to a male not related to JonBenet.
This convinced prosecutors that the Ramsey family could not have been responsible for JonBenet's death.
After the creation of a DNA profile, what's next? It depends on the intended purpose of the DNA profile.
If the DNA comes from samples recovered in a criminal investigation, prosecutors in the United States will enter it into CODIS, the Combined Data Index System. CODIS is a computer program maintained by the FBI , which operates databases across the country.
These databases contain more than five million profiles. CODIS contains several different indexes:
- The Offender Index contains the profiles of people convicted of various crimes. The crimes that result in inclusion in the Offender Index vary depending on the state, and they range from certain misdemeanors to sexual offenses and murder.
- The Arrestee Index contains profiles of people arrested for committing specific violent felonies. The exact crimes also vary by state.
- The Forensic Index contains profiles taken from crime scene evidence, including blood , saliva, semen and tissue.
- The Missing Persons Index consists of two indexes: Unidentified Persons, which contains the profiles recovered from the remains of unidentified persons, and Reference, which contains profiles of relatives of missing persons. These two indexes are periodically compared to each other to determine if a missing person's remains have been recovered.
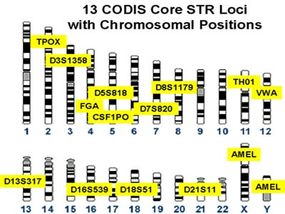
CODIS uses algorithms to compare 13 different STR locations, plus one that determines the gender of the person in question. It has rules and safeguards to protect the privacy of people whose profiles are in the database.
The matching algorithms — which an analyst must confirm — can produce leads for law enforcement or even identify a potential assailant. The downside of using CODIS is that it's only as strong as the number of profiles included, and there are more than one million profiles that are not yet included.
Prosecutors can also use DNA experts to match profiles while building cases where there's a high degree of certainty of the assailant.
Profiling Biological Evidence for Past Convictions
DNA profiling is being used more and more for people convicted before its common use, which began in the late 1980s. Since the early 1990s, convicted criminals have been able to use the latest DNA profiling technology as part of their appeals process.
Most states have laws explicitly describing the rights convicted criminals have to DNA testing. In some cases, people can request additional testing anytime, while in others, they must do so within a few years of their conviction.
Attention to post-conviction DNA testing began with a 1996 National Institute of Justice report that spotlighted 28 people convicted of rape and murder who had been exonerated due to later DNA testing.
Since 1989, more than 218 convicted criminals have been released after DNA testing proved their innocence. The true perpetrator was identified in 84 of those cases [source: The Innocence Project].

Aside from criminal trials and appeals, DNA profiling has become an important tool in genealogy. Many companies provide DNA profiling for this purpose. One of the largest ones, Family Tree DNA, uses Y-SRT testing to determine paternal lineage and mtDNA (mitochondrial DNA testing) to determine maternal lineage.
When an embryo is conceived, its mitochondria — structures within cells that convert energy from food — come from the mother's egg cell, whereas the father's sperm contributes only nuclear DNA [source: Human Genome Project , U.S. National Library of Medicine ].
National DNA Database
The creation and storage of DNA profiles are also very controversial. As the databases searched by CODIS have expanded to include profiles of more than just convicted criminals, some people have begun to worry about what law enforcement, the government or even private companies may be able to do with the information.
Once your profile is in a database, you need a court order to get it removed. If you're using a private database for the purposes of genealogy, however, you can request the removal of your profile.
In April 2008, the Genetic Information Discrimination Act became law. The act keeps insurance companies and employers from discriminating against people who may be genetically predisposed to a disease.
The profiles vary in the amount of detail they can provide and in how far back in your ancestry they can determine a match. A Y-DNA67, for example, can show an extremely close connection between ancestors. It tests the Y chromosome for genetic matches between males.
A perfect match of 67 markers on each person's DNA strand means they have a common ancestor in recent history [source: Family Tree DNA ]. Family Tree DNA maintains databases of people looking for ancestors, and when it finds a match, it notifies both parties.
So DNA profiling can be very useful, but how accurate is it in determining a match? Family Tree DNA says it's the "most comprehensive DNA matching database in the industry" because more than 2 million people have used the service. However, DNA profiling, especially in criminal cases, isn't infallible.
DNA Profiling vs. Genetic Testing
Although DNA profiling can reveal ancestry, companies that specialize in them don't perform any kind of testing specifically to trace hereditary defects or disease.
However, genetic testing, which involves more than just DNA profiling, helps reveal hereditary predispositions to some diseases and birth defects. During genetic testing, DNA is profiled and analyzed along with RNA, proteins and other factors.
When DNA profiling first came to use in criminal cases, it was often difficult for prosecutors and defense attorneys, as well as the experts they hired to testify, to explain the significance of their DNA match to the jury.
Fingerprints are still an ironclad way to identify someone, but an expert testifying about fingerprints discusses them in terms of "points of similarity." An expert discusses DNA matches in terms of statistical probability using what is currently known about DNA similarity within the general population . This often confused the jury or they interpreted it incorrectly.

For example, an expert testifying about DNA profiling for the prosecution might say that the DNA profile created from the crime scene evidence has a 4-to-5 probability (or 80 percent chance) of matching the DNA profile created from the defendant's sample.
Saying that the probability of the match is 80 percent, however, is not the same thing as saying that the probability of the accused person's guilt is 80 percent.
On the other hand, an expert testifying about DNA profiling for the defense could say something like, "The likelihood that this person's DNA was found at the crime scene, but he did not commit the crime, is 1 in 10 (or 10 percent)."
That isn't a very high probability, but it doesn't take into account the fact that the accused isn't some random person plucked off the street. It's not likely that the DNA profile is the only reason why he or she was arrested for the crime; DNA is just one piece in a very large puzzle.
Margin of Error
DNA profiling and its interpretation have come under fire. RFLP analysis was in part discontinued because of the possibility for error.
The risk of a coincidental match using RFLP is 1 in 100 billion. However, in laboratory settings, this risk is probably higher because technicians may misread similar patterns as identical or otherwise perform the analysis incorrectly.
A 2002 study of the accuracy of DNA laboratories in the United States conducted by the University of Texas showed that 1 in 100 profiles may give a false result.
STR analysis is not as subjective, but any DNA profile can give a false result if it ends up contaminated. Although there have been no documented cases of a laboratory worker intentionally contaminating a DNA sample, DNA samples have been contaminated or even faked by criminals to avoid prosecution.
Manipulating Evidence
In 1992, a patient of Dr. John Schneeberger accused him of sexually assaulting her while she was sedated. A DNA profile was created using the sample that he left on the victim. A profile from a sample of his blood did not match the crime scene sample, and the case was closed.
The victim persisted, and eventually, Dr. Schneeberger was convicted after additional DNA samples showed a match. He was able to avoid the initial match by implanting a drain in his arm filled with another man's blood and an anticoagulant, and skillfully getting the technician who drew his blood to do so from that spot.
Ultimately, DNA profiling has proved to be an amazing tool. However, it's just one of the many tools used to find the truth in criminal investigations, genealogy searches and testing for disease. There is rarely a 100 percent certainty of anything.
Lots More Information
Related howstuffworks articles.
- How DNA Works
- How DNA Evidence Works
- How Blood Works
- How Fingerprinting Works
- How Biometrics Works
- How the FBI Works
- How the CIA Works
- How the ACLU Works
- Baden, Michael. "DNA Profiling." http://www.kathyreichs.com/dnaprofiles.htm (July 23, 2008.)
- Baetke, James. "Ramsey breakthrough comes via 'touch DNA'." The Daily Camera, July 9, 2008. http://www.dailycamera.com/news/2008/jul/09/ramsey-breakthrough-comes-touch-dna/
- Billings, P.R., editor. "DNA on Trial: Genetic Identification and Criminal Justice." Cold Spring Harbor Laboratory Press, 1992.
- Brinton, Kate and Kim-An Lieberman. "Basics of DNA Fingerprinting." Biology Department, University of Washington, May 1994. http://protist.biology.washington.edu/fingerprint/dnaintro.html
- "Comparison of State Post-conviction DNA Laws." National Conference of State Legislatures, 2008. http://www.ncsl.org/programs/health/genetics/DNAchart.htm
- Evans, Colin. "The Casebook of Forensic Detection." Penguin Books Ltd, 2007.
- Groleau, Rick. "Create a DNA Fingerprint." NOVA Online, November 2000. http://www.pbs.org/wgbh/nova/sheppard/analyze.html
- Innocence Project. http://www.innocenceproject.org/
- Kirby, Lorne T. "DNA Fingerprinting: An Introduction." W.H. Freeman and Company, 1992.
- Lawless, Jill. "DNA fingerprinting sparks fresh worries." MSNBC, September 8, 2002. http://www.msnbc.msn.com/id/5944270/
- Lee, Henry C. and Frank Tirnady. "Blood Evidence: How DNA is Revolutionizing the Way We Solve Crimes." Perseus Publishing, 2003.
- Manning, Lona. "Rapist, M.D." Crime Magazine, February 6, 2004. http://www.crimemagazine.com/03/rapistmd,0403.htm
- Walsh, Nick Patton. "False result fear over DNA tests." The Observer, January 27, 2002. http://www.guardian.co.uk/uk/2002/jan/27/ukcrime.research
- "What is 'touch' DNA?" CNN.com/Crime, 2008. http://www.cnn.com/2008/CRIME/07/09/touch.dna.ap/index.html
Please copy/paste the following text to properly cite this HowStuffWorks.com article:
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Research Briefing
- Published: 09 May 2024
Quantitative profiling of regulatory DNA activity at single-cell resolution
Nature Methods ( 2024 ) Cite this article
990 Accesses
34 Altmetric
Metrics details
- Gene regulation
We developed a two-pronged strategy to functionally probe the enormous repertoire of noncoding DNA within genomes. Our approach markedly improved signal-to-noise ratio and successfully intersected single-cell genomics with reporter assays. The result delivers a multiplex and highly quantitative readout of regulatory sequences’ activity in dynamic and multicellular systems.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
The ENCODE Project Consortium. et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583 , 699–710 (2020). An overview article presenting the vast catalogue of regulatory elements in mammalian genomes.
Article CAS Google Scholar
Patwardhan, R. P. et al. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 27 , 1173–1175 (2009). This paper reports the massively parallel reporter assay strategy to profile the function of regulatory DNA at scale.
Article CAS PubMed PubMed Central Google Scholar
Litke, J. L. & Jaffrey, S. R. Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat. Biotechnol. 37 , 667–675 (2019). This paper introduces the Tornado system to stabilize short polymerase III-driven RNAs.
Zhao, S. et al. A single-cell massively parallel reporter assay detects cell-type-specific gene regulation. Nat. Genet. 55 , 346–354 (2023). This paper reports an assay that combines scRNA-seq with multiplex reporters.
Mangan, R. J. et al. Adaptive sequence divergence forged new neurodevelopmental enhancers in humans. Cell 185 , 4587–4603.e23 (2022). This paper presents another example of an assay that combines scRNA-seq with STARR-seq.
Download references
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This is a summary of: Lalanne, J.-B. et al. Multiplex profiling of developmental cis- regulatory elements with quantitative single-cell expression reporters. Nat. Methods https://doi.org/10.1038/s41592-024-02260-3 (2024).
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Quantitative profiling of regulatory DNA activity at single-cell resolution. Nat Methods (2024). https://doi.org/10.1038/s41592-024-02261-2
Download citation
Published : 09 May 2024
DOI : https://doi.org/10.1038/s41592-024-02261-2
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing: Translational Research newsletter — top stories in biotechnology, drug discovery and pharma.

- Open access
- Published: 10 May 2024
Machine learning based DNA melt curve profiling enables automated novel genotype detection
- Aaron Boussina 1 ,
- Lennart Langouche 2 ,
- Augustine C. Obirieze 2 ,
- Mridu Sinha 3 ,
- Hannah Mack 3 ,
- William Leineweber 3 ,
- April Aralar 3 ,
- David T. Pride 4 ,
- Todd P. Coleman 5 &
- Stephanie I. Fraley 3
BMC Bioinformatics volume 25 , Article number: 185 ( 2024 ) Cite this article
125 Accesses
Metrics details
Surveillance for genetic variation of microbial pathogens, both within and among species, plays an important role in informing research, diagnostic, prevention, and treatment activities for disease control. However, large-scale systematic screening for novel genotypes remains challenging in part due to technological limitations. Towards addressing this challenge, we present an advancement in universal microbial high resolution melting (HRM) analysis that is capable of accomplishing both known genotype identification and novel genotype detection. Specifically, this novel surveillance functionality is achieved through time-series modeling of sequence-defined HRM curves, which is uniquely enabled by the large-scale melt curve datasets generated using our high-throughput digital HRM platform. Taking the detection of bacterial genotypes as a model application, we demonstrate that our algorithms accomplish an overall classification accuracy over 99.7% and perform novelty detection with a sensitivity of 0.96, specificity of 0.96 and Youden index of 0.92. Since HRM-based DNA profiling is an inexpensive and rapid technique, our results add support for the feasibility of its use in surveillance applications.
Peer Review reports
Introduction
Effective large-scale monitoring and surveillance of novel pathogens are critical components of contemporary public health strategy [ 1 , 2 ]. Genotypic screening of pathogens, in particular, enables researchers and clinicians to gain nuanced insights into disease transmission patterns, virulence factors, antibiotic resistance profiles, and potential outbreak sources [ 3 ]. Modern techniques for genotypic screening, however, can be cost-prohibitive, slow, and intractable to scale [ 4 ]. High Resolution Melting (HRM) analysis offers an emerging alternative that enables rapid, effective, and economical post-amplification nucleic acid characterization for profiling DNA sequences [ 5 , 6 , 7 , 8 , 9 , 10 , 11 ].
HRM involves the use of a DNA-binding dye added to DNA samples, which fluoresces upon intercalating into the double-stranded structure. As the sample is heated, the DNA denatures, resulting in a loss of fluorescence which is recorded to produce a melt curve. Recent advancements have positioned HRM as not just a check on amplification product homogeneity, but also as a reliable method for heterozygote detection [ 5 , 12 ]. Moreover, with improvements in heat transfer and reaction engineering, homozygous melt curves can now be leveraged as sequence-specific signatures [ 13 , 14 , 15 , 16 ].
When applied to the problem of identifying and differentiating bacteria, universal primers have been used to target the 16S rRNA gene, which is highly conserved in bacteria but contains variable regions that are specific to different species or strains. This enables broad-based amplification of the bacterial 16S gene with universal primers, while relying on melt to genotype the hypervariable sequences specific to organism identity. The use of machine learning (ML) classification, where each unique sequence-specific curve signature represents a pathogen class, offers a principled framework to utilize HRM as a broad-based sequence profiling tool. This could be especially valuable in clinical diagnostics, where specifically identifying pathogenic bacteria is crucial for determining the appropriate treatment [ 17 , 18 , 19 ].
The growth in the amount of HRM data available for ML training has further accelerated the practical application of these methods for pathogen profiling. The rising prominence of digital PCR has led to the evolution from traditional HRM to digital HRM (dHRM) which enables 200-fold increases in the number of melt curves [ 20 , 21 ]. This methodology originated from our prior work in which we introduced a unique dHRM platform that employs specialized heat transfer and imaging mechanisms to simultaneously melt thousands of digital PCR (dPCR) reactions [ 13 , 14 , 22 ]. A distinct feature of this methodology is its digital design, in which each reaction is characterized by the presence or absence of a genome as its DNA template. While traditional HRM might typically operate on a 96-well plate, our dHRM technique utilizes a dPCR chip with 20,000 partitions, resulting in 20,000 HRM curves.
Existing literature has demonstrated effective application of Naïve Bayes (NB) [ 23 ], Support Vector Machines (SVM) [ 15 , 16 ], k-Nearest Neighbors using Dynamic Time Warping [ 24 ], Random Forest (RandF) [ 25 , 26 ], and Neural Networks [ 27 ], for the purpose of pathogen classification using melt curve data. SVM algorithms have shown notable performance even with relatively few melt curves. However, in the traditional one-versus-all application for multiclass classification, these methods are ill-suited to address melt curves that lie outside the distribution of their training set. Specifically, when presented with an out-of-distribution melt curve, these classifiers may erroneously classify the signature of an emerging pathogen as a known pathogen class. Thus, to enable broad-based pathogen surveillance, there is substantial promise in ML that can accurately identify the emergence of novel genotypes.
In this work, we leverage the massive datasets generated from our dHRM platform and broad-based 16S gene amplification strategy to evaluate the performance of common ML classifiers in the identification of novel and known bacterial genotypes. To our knowledge, this work represents the first study in which multiple broadly applicable ML classifiers have been investigated for the purpose of novelty detection with HRM. Previous work by Andini et al. utilized Naïve Bayes and a custom distance metric based on the Hilbert Transformation, but this approach was limited in efficacy since it aligned melt peaks to a single temperature [ 23 ]. We hypothesize that by incorporating a larger HRM dataset than previously reported, extracting significant geometric features of the melt curves, selecting the optimal ML algorithm, and developing an appropriate metric of model confidence, we can more accurately automate the identification of novel genotypes. We test this hypothesis using both experimental samples and simulated melt curves generated using the uMelt tool [ 28 ]. We focus on the specific use-case of bacteremia in neonates in which a small number of bacterial organisms are typically implicated, but emerging and opportunistic infections can occur [ 29 ].
Material and methods
Bacterial strains.
The bacterial species used in this study and their corresponding melt curves are described in Additional file 1 : Table S1. These bacteria are the primary causative pathogens in cases of neonatal sepsis [ 29 , 30 ]. We obtained isolates from Dr. David Pride (University of California San Diego School of Medicine) as well as the American Tissue Culture Collection (ATCC, Old Town Manassas, VA). Bacteria were cultured in Lurie-Bertani (LB) broth or Tryptic Soy broth (TSB), as required, and incubated overnight at 37 °C.
Bacterial genomic DNA extraction and PCR
Following culturing, we performed DNA extraction using Wizard Genome DNA Purification kit (Promega Corporation, Madison, WI). We assessed the quality and concentration of the extracted DNA using spectrophotometric absorbance measurements and confirmed the identities of the species from sequencing. We prepared genomic DNA dilutions for use with dPCR and used the commercially available QuantStudio 3D Digital PCR 20 K chip v2 (Applied Biosystems, Foster City, CA) for amplification. We followed the manufacturer’s recommended process, but customized our reagents. The composition of our dPCR master mix is described in our prior work. It includes 1 μL of sample, 0.15 μM forward primer 5′-GYGGCGNACGGGTGAGTAA-3′ (Integrated DNA Technologies, Coralville, IA), 0.15 μM reverse primer 5′-AGCTGACGACANCCATGCA-3′ (Integrated DNA Technologies, Coralville, IA), 0.02 U/μL of Phusion HotStart Polymerase (Thermo Fisher Scientific, Waltham, MA), 0.2 mM dNTPs (Invitrogen, Carlsbad, CA), 1X Phusion HF Buffer containing 1.5 mM MgCl2 (Thermo Fisher Scientific, Waltham, MA), 2.5X EvaGreen (Biotium, Fremont, CA), 2X ROX (Thermo Fisher Scientific, Waltham, MA), and ultrapure PCR water (Quality Biological Inc., Gaithersburg, MD) to bring the total volume to 14.5 μL. We loaded the chip by spreading 14.5µL of the master mix per the manufacturer’s recommendation. We then cycled the the dPCR on a flatbed thermocycler with the following cycle settings: an initial enzyme activation (98 °C, 30 s), followed by 70 cycles (95 °C, 30 s, 59 °C, 30 s, 72 °C, 60 s).
DNA melt curve generation and preprocessing
The architecture of our U-dHRM device has been previously described [ 13 , 14 ]. A copper plate hosts the microfluidic dPCR chip, separated by a thin layer of thermal grease to ensure efficient heat transfer. Temperature control is achieved through a thermoelectric module (TE Technology Inc., Traverse City, MI), PID controller (Meerstetter Engineering GmbH, Rubigen, Switzerland), Class 1/3B resistance temperature detector (RTD) (Heraeus, Hanau, Germany) embedded in the copper block, K-type thermocouple (OMEGA Engineering, Stamford, CT), and heat sink. We secure the device on-stage for optimal fluorescent imaging using a custom-made adapter. A Nikon Eclipse Ti microscope (Nikon, Tokyo, Japan) captures simultaneous fluorescent images from heat ramping the DNA-intercalating dye, EvaGreen (Ex/Em: 488 nm/561 nm) and the control dye, ROX (Ex/Em: 405 nm/488 nm). An automated image processing algorithm implemented in MATLAB is used to generate the melt curves. We perform background subtraction using the linear method described in [ 31 ]. This horizontally aligns the tails of the melt curves with the x-axis, to ensure they are most similar to the theoretically predicted uMelt curves.
Feature engineering
The resulting preprocessed melt curves contain fluorescence loss values (−dF/dT) for 410 temperature steps in the range [51 °C, 92 °C]. We model this as a 1-dimensional time-series and apply a signature transform to each melt curve. The signature method is a non-parametric feature extractor that computes a series of integrals along a data path that fully capture its order and area [ 32 , 33 ]. The signature method is optionally time-shift invariant and is sensitive to the geometric shape of the path. We apply the signature transform on a rolling window with a kernel size of 20 and a stride of 8 across the time-series with time and basepoint augmentations and a signature depth of 3 to generate a set of features for the downstream classification tasks.
Machine learning model selection
We set out to compare five ML methods: Logistic Regression, Naïve Bayes (NB), Support Vector Machines (SVM), Neural Networks and Random Forest (RandF). To address correlation between the input signature features, we used L2 regularization for the logistic regression, SVM, and neural network models. At the end of this work we briefly discuss how calibrating the probabilities affects the results. We built and implemented all algorithms using the scikit-learn package within Python programming language [ 34 ]. All data and code are available on https://github.com/aboussina/dHRM-novelty-detection .
Quantification of genotypic differences
To further assess the utility of our derived HRM signatures for the identification of distinct genotypes, we analyzed the ability of the aforementioned ML models to quantify the degree of genotypic differences between species. That is, beyond simply classifying a melt curve to a given species, we reformulated this as a regression problem where the target variable is a metric for genotypic difference. We calculated this metric by mapping our ten bacterial species on the SILVA phylogenetic tree and computing the patristic distance between the node of each species and the E. coli node [ 35 ]. We evaluated the performance of this regressor using the c-statistic as described in [ 36 ].
Leave-one-group-out (LOGO) experiments
We evaluated the capability of these machine learning models for the task of identifying novel melt curves, i.e. those belonging to species unrepresented in the training set, using a leave-one-group-out experimental design. For each of our ten bacterial species, we held out its melt curves and split the curves of the remaining nine species into training and test sets (80:20 ratio). Then, the held-out species' curves were added to the test set. The machine learning model was trained on the training set and then tested on its ability to recognize the curves of the held-out species as novel within the modified test set. This process was repeated for each of the ten species. The schematic of this approach is presented in Fig. 1 . To measure our model's efficacy in novelty detection, we used Youden’s index, a metric that assesses the performance of a binary diagnostic test.

Workflow for ML novelty detection
Threshold selection
We set the threshold for novelty identification on a single LOGO experiment to the Youden Index (i.e. the point on an ROC curve at which sensitivity + specificity − 1 is maximized). However, to practically identify novel organisms, the ideal threshold should apply across all ten LOGO experiments. Thus, we calculate a ‘practical’ threshold by accumulating all the LOGO experiments and determining the Youden Index from their combined ROC curve. We then assess the performance of each LOGO experiment using this combined ‘practical’ threshold. We investigated applying sample weights to each experiment based on the number of left-out curves, but didn’t observe a significant change in the selected threshold (data not shown).
Threshold validation with synthetic melt curves
We generated in-silico melt curves using uMelt for 50 clinically relevant bacterial pathogens, including category A and B biothreat agents and their surrogates from [ 37 ] (Additional file 1 : Table S1). We added real dHRM noise to these synthetic melt curves to more realistically capture sample variation as described [ 26 ]. We created 100 melt curves per species, with a unique noise residual applied to each individual melt curve.
Preprocessing
Figure 2 and Additional file 1 : Figs. S1–S2 show the results of background subtraction on the experimental melt curves. Also shown are the simulated melt curves from uMelt with added dHRM noise. Supplemental Additional file 1 : Fig. S3 shows an overview of the synthetically created melt curves for all 50 pathogens. As demonstrated, the background subtraction effectively aligns the curves with the x-axis. Further, the simulated curves show similar characteristics to the experimental observations.

Overview of dHRM datasets. A Experimentally obtained dHRM melt curves. B Ten examples of synthesized melt curves using a combination of uMelts and real dHRM melt curve noise. Full and short melt refer to using the entire length of the melt curve or a shorter window around the melt peak location
Classification and regression performance
The ML methods show very similar classification accuracy, which is summarized in Table 1 . No significant difference can be observed between the ‘full melt’ and ‘short melt’ classification results, which implies there might not be any additional information in the tail of the melt curve. Additional file 1 : Figure S4 shows the correlation between melt curve distance (defined as the average pointwise distance from the average E. coli curve) and patristic distance from E. coli . We observe modest correlation with a couple of distinct outliers. The performance of a select ML method (RandF, n = 500) to quantify the patristic distance is listed in Additional file 1 : Table S2. We observe strong performance (c-statistic: 0.96) for this regression task indicating that the derived features enable quantification of the genotypic differences between organisms.
Leave-one-group-out (LOGO) experiments and threshold selection
Figure 3 shows the results of one LOGO experiment for one ML method (RandF, n = 500). The optimal threshold is found by plotting the ROC curve (Fig. 3 B) and selecting its Youden index.

Leave-one-species-out cross validation to determine probability threshold. A Boxplot of the classification probabilities of each of the ten species. In this experiment C. koseri is the left-out species, which means it is left out of the training set and added to the test set. This experiment is repeated for each of the ten species, and an optimal threshold can be found for each of them. This experiment is repeated for all ML methods (method shown here is RandF (n = 500)). B ROC curve that is used to find the optimal threshold. Youden’s Index is chosen as the optimal threshold, it is the point on the ROC curve where sensitivity + specificity − 1 is maximized
Figures 4 and 5 summarize the results of the accumulated LOGO experiments. The ROC curves of the 10 individual LOGO experiments as well as the accumulated ROC curve are shown in Fig. 4 . The performance, as measured by Youden’s index is shown as a function of classification method in Fig. 5 . Each bar shows the average performance across ten LOGO experiments. ‘Optimal thresholds’ means selecting the best threshold for each LOGO experiment individually. ‘Practical threshold’ means selecting the optimal threshold for the accumulated LOGO experiments and applying it to all LOGO experiments separately. Random Forest outperforms the other methods, but Neural Nets and SVMs still perform relatively well.

Accumulating the leave-one-group-out (LOGO) experiments results in a ‘practical’ threshold. A ROC curve for all ten LOGO experiments accumulated with RandF (n = 500). The optimal threshold is again found by Youden’s Index. We have named it the ‘practical’ threshold as one threshold has to be chosen (as opposed to a separate threshold for each LOGO experiment) when further validating the model on unseen ‘novel’ melt curves. It is the optimal threshold for all ten LOGO experiments combined. B Choosing a practical threshold implies that each LOGO experiment individually will be performing at a suboptimal threshold, which translates to a suboptimal operating point on the ROC curve

Summary of LOGO novelty detection results. Average novelty detection performance across ten species measured by Youden’s index as a function of classification method. Optimal means selecting the best threshold for each leave-one-species-out experiment. Practical means selecting one threshold and applying it to all leave-one-species-out experiments
Threshold validation and novelty detection
Figure 6 shows boxplots and ROC curves used to validate the previously obtained ‘practical’ threshold using the signature features (Fig. 6 A, B ) as well as the raw melt curves (Fig. 6 C, D ). The method shown in Fig. 6 is RandF (n = 100), which was one of the best performing methods, similar figures for all other methods are available in the supplementary data (Additional file 1 : Fig. S5). When the practical threshold is close to the optimal threshold, the practical operating point on the ROC curve (Fig. 6 B) will be close to the optimal one (Youden’s index). When this is the case, it confirms that our proposed method for obtaining a practical threshold for novelty detection, is indeed valid. Both feature sets (signatures and the raw melt curves) achieve strong discriminative performance of novel organisms based on the model score. The use of signature features enables an overall slight improvement but notably results in reduced novelty detection of T. pallidum. Figure 7 A shows an overview of the results for all ML methods. The average difference between the optimal and practically attained Youden index across the ten ML methods is just 0.019 with a standard deviation of 0.025. This serves as a confirmation of our threshold selection process using the accumulated LOGO experiments. Random Forest and SVM (rbf) perform the best, with the Neural Nets a close third.

Validation of practical threshold on synthesized set of melt curves. A The practical threshold, selected through the LOGO experiments, was validated on the signature features from a new dataset consisting of 50 species, each with 100 synthetic melt curves with real dHRM noise. The performance of E. coli was confirmed to be an outlier using one-tailed t-tests. B ROC curve. When the practical threshold is close to the optimal threshold, it serves as a validation for the threshold selection process. C The practical threshold, repeated on the raw experimental and synthetic melt curves. D ROC curve using the raw melt curves. The ML method shown here is RandF (n = 100)

Summary of practical threshold validation results. A Average novelty detection performance on 50 unseen species measured by Youden’s index as a function of classification method. B Dropping E. coli , an outlier group, results in improved performance for almost all methods (results including E. coli are overlayed in gray). SVM (rbf) is more robust against this outlier behavior and sees less improvements
Further improvements
We observed that the classification probabilities for the E. coli group of melt curves are more spread out (Fig. 6 A) and might be an outlier group compared to the other species. This was apparent for multiple ML methods and was confirmed with one-tailed t-tests (e.g. for the short melt curves: P < 0.01 for Logistic Regression, Gaussian NB, SVM (rbf and linear), Neural Net (identity and relu) and RandF (n = 100 and n = 500)). As a result of this, we ran all steps again, but this time leaving out E. coli , to see if we could further optimize our novelty detection method. Figure 7 B shows that results do indeed improve when leaving E. coli out. Random Forest and SVM (rbf) are the top performers, and their results are further summarized in Table 2 . The best performance achieved is a Youden index of 0.93, corresponding to a specificity of 0.97 and sensitivity of 0.96.
We also investigated whether calibrating the probabilities using scikit-learn’s ‘CalibratedClassifierCV’ function would improve the outcome. We tested both the ‘sigmoid’ method, which corresponds to Platt’s method (i.e., a logistic regression model) or the ‘isotonic’ method, which is a non-parametric approach. Results are summarized in supplemental Additional file 1 : Fig. S6. As expected, we see a large improvement for Naïve Bayes. We also see a significant improvement for Logistic Regression. None of the calibrated methods outperform the best results (SVM, Neural Net, RandF) as outlined in Fig. 7 and Table 2 though.
Our work demonstrates the utility of time-series classification algorithms in resolving multiple bacterial organism melt curves, and in identifying previously unknown (novel) melt curves that are not represented in the database. The large amount of dPCR chip-generated melt curve data enabled the development of machine learning classifiers and novelty detection algorithms, which distinguishes this study from previous studies which did not assess out-of-distribution data and utilized small datasets of melt curves [ 38 , 39 ].
The only other published method specifically aimed at melt curve novelty detection [ 23 ] aligns the melt curves to one specific temperature, losing the useful melt peak location information in the process. We have selected the most widely used ML methods in HRM analysis and have shown that they are all able to classify our dHRM database with very high accuracy. Interestingly, some drastically outperform others when it comes to novelty detection. We find that Neural Nets, SVMs, and Random Forest outperform the other ML methods, even after calibrating the probabilities. Random forests utilizing features extracted from a time-series have been shown to perform well on time-series classification tasks [ 40 ]. Here, we show that its well-calibrated probabilities are also particularly useful for conducting HRM novelty detection.
The performance of our approach was improved with the removal of data for the outlier species E. coli . One reason for the lower performance of E. coli compared to the other species could be that is has the lowest number of melt curves available (Additional file 1 : Table S1), which results in a smaller amount of training data available for the ML methods. We do not expect E. coli to inherently have more heterogeneity in its sequence compared to other species. Melt curve shape variance might be another contributor to its outlier behavior as it has the third most variance in shape (from the ten species) as measured by dynamic time warping (DTW), in our previous work [ 26 ].
No major differences were found between using the full length and short version of the melt curves, although for most methods the short version does outperform the full length, showing that there might not be any additional information in the low-temperature tail of the curve, and including it might even confound the novelty detection performance.
There are several limitations to our work. First, while we benchmarked novelty detection across a suite of machine learning algorithms, we did not perform hyperparameter tuning. It is possible that approaches such as Bayesian optimization could improve novelty detection performance. Further, we did not utilize any distance-based metric for evaluating out-of-distribution novel organisms; opting instead to leverage the output scores from discriminative classifiers to select an optimal threshold. Our approach provides an effective way to incorporate novelty detection within a large machine learning framework, but future work is required to evaluate alternative distance-aware methodologies [ 41 ].
In conclusion, advances in machine learning and ‘big data’ generation are opening up more opportunities for the advancement of HRM, which due to its speed, low cost, and simplicity was already attractive. The opportunity to use HRM as a discovery tool as well as profiling technology will further advance HRM technology towards its application in research and clinical diagnostics.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. Bacterial 16S gene sequences were sourced from the National Center for Biotechnology Information Reference Sequence Database located at https://www.ncbi.nlm.nih.gov/refseq/ .
Tacconelli E. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development. World Health Organization; 2017.
Heymann DL, Shindo N. COVID-19: What is next for public health? The lancet. 2020;395(10224):542–5.
Article CAS Google Scholar
Didelot X, Bowden R, Wilson DJ, Peto TE, Crook DW. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet. 2012;13(9):601–12.
Article CAS PubMed PubMed Central Google Scholar
Kwong JC, McCallum N, Sintchenko V, Howden BP. Whole genome sequencing in clinical and public health microbiology. Pathology. 2015;47(3):199–210.
Article CAS PubMed Google Scholar
Reed GH, Wittwer CT. Sensitivity and specificity of single-nucleotide polymorphism scanning by high-resolution melting analysis. Clin Chem. 2004;50(10):1748–54.
Liew M, Pryor R, Palais R, Meadows C, Erali M, Lyon E, et al. Genotyping of single-nucleotide polymorphisms by high-resolution melting of small amplicons. Clin Chem. 2004;50(7):1156–64.
Bidet P, Liguori S, Plainvert C, Bonacorsi S, Courroux C, d Humières C, et al. Identification of group A streptococcal emm types commonly associated with invasive infections and antimicrobial resistance by the use of multiplex PCR and high-resolution melting analysis. Eur J Clin Microbiol Infect Dis. 2012;31(10):2817–26.
Roth AL, Hanson ND. Rapid detection and statistical differentiation of KPC gene variants in Gram-negative pathogens by use of high-resolution melting and ScreenClust analyses. J Clin Microbiol. 2013;51(1):61–5.
Zianni MR, Nikbakhtzadeh MR, Jackson BT, Panescu J, Foster WA. Rapid discrimination between Anopheles gambiae s.s. and Anopheles arabiensis by High-Resolution Melt (HRM) analysis. J Biomol Tech. 2013;24(1):1–7.
PubMed PubMed Central Google Scholar
Pritt BS, Mead PS, Johnson DKH, Neitzel DF, Respicio-Kingry LB, Davis JP, et al. Identification of a novel pathogenic Borrelia species causing Lyme borreliosis with unusually high spirochaetaemia: a descriptive study. Lancet Infect Dis. 2016;16(5):556–64.
Article PubMed PubMed Central Google Scholar
Langouche L. Advancing rapid infectious disease screening using a combined experimental/computational approach. San Diego: University of California; 2021.
Google Scholar
Cheng J-C, Huang C-L, Lin C-C, Chen C-C, Chang Y-C, Chang S-S, et al. Rapid detection and identification of clinically important bacteria by high-resolution melting analysis after broad-range ribosomal RNA real-time PCR. Clin Chem. 2006;52(11):1997–2004.
Velez DO, Mack H, Jupe J, Hawker S, Kulkarni N, Hedayatnia B, et al. Massively parallel digital high resolution melt for rapid and absolutely quantitative sequence profiling. Sci Rep. 2017;8(7):42326.
Article Google Scholar
Sinha M, Mack H, Coleman TP, Fraley SI. A high-resolution digital DNA melting platform for robust sequence profiling and enhanced genotype discrimination. SLAS Technol. 2018;23(6):580–91.
Article PubMed Google Scholar
Athamanolap P, Parekh V, Fraley SI, Agarwal V, Shin DJ, Jacobs MA, et al. Trainable high resolution melt curve machine learning classifier for large-scale reliable genotyping of sequence variants. PLoS ONE. 2014;9(9): e109094.
Fraley SI, Athamanolap P, Masek BJ, Hardick J, Carroll KC, Hsieh Y-H, et al. Nested machine learning facilitates increased sequence content for large-scale automated high resolution melt genotyping. Sci Rep. 2016;18(6):19218.
Aralar A, Goshia T, Ramchandar N, Lawrence SM, Karmakar A, Sharma A, Sinha M, Pride DT, Kuo P, Lecrone K, Chiu M, Mestan KK, Sajti E, Vanderpool M, Lazar S, Crabtree M, Tesfai Y, Fraley SI. Universal digital high-resolution melt analysis for the diagnosis of bacteremia. J Mol Diagn. 2024;26(5):349–63.
Goshia T, Aralar A, Wiederhold N, Jenks JD, Mehta SR, Karmakar A, E.S. M, Sharma A, Sun H, Kebadireng R, White PL, Sinha M, Hoenigl M, Fraley SI. 0. Universal digital high-resolution melting for the detection of pulmonary mold infections. J Clin Microbiol. e01476–23.
Sinha M, Jupe J, Mack H, Coleman TP, Lawrence SM, Fraley SI. Emerging Technologies for Molecular Diagnosis of Sepsis. Clin Microbiol Rev. 2018;31(2):e00089-17.
Athamanolap P, Hsieh K, O’Keefe CM, Zhang Y, Yang S, Wang T-H. Nanoarray digital polymerase chain reaction with high-resolution melt for enabling broad bacteria identification and pheno-molecular antimicrobial susceptibility test. Anal Chem. 2019;91(20):12784–92.
Rolando JC, Jue E, Barlow JT, Ismagilov RF. Real-time kinetics and high-resolution melt curves in single-molecule digital LAMP to differentiate and study specific and non-specific amplification. Nucleic Acids Res. 2020;48(7): e42.
Aralar A, Yuan Y, Chen K, Geng Y, Ortiz Velez D, Sinha M, et al. Improving quantitative power in digital PCR through digital high-resolution melting. J Clin Microbiol. 2020;58(6):66.
Andini N, Wang B, Athamanolap P, Hardick J, Masek BJ, Thair S, et al. Microbial typing by machine learned DNA melt signatures. Sci Rep. 2017;6(7):42097.
Lu S, Mirchevska G, Phatak SS, Li D, Luka J, Calderone RA, et al. Dynamic time warping assessment of high-resolution melt curves provides a robust metric for fungal identification. PLoS ONE. 2017;12(3): e0173320.
Bowman S, McNevin D, Venables SJ, Roffey P, Richardson A, Gahan ME. Species identification using high resolution melting (HRM) analysis with random forest classification. Aust J Forensic Sci. 2017;25:1–16.
Langouche L, Aralar A, Sinha M, Lawrence SM, Fraley SI, Coleman TP. Data-driven noise modeling of digital DNA melting analysis enables prediction of sequence discriminating power. Bioinformatics. 2020;6:66.
Adelman JD, McKay WR, Lillis J, Lawson K. High-resolution melt curve classification using neural networks.
Dwight Z, Palais R, Wittwer CT. uMELT: prediction of high-resolution melting curves and dynamic melting profiles of PCR products in a rich web application. Bioinformatics. 2011;27(7):1019–20.
Klinger G, Levy I, Sirota L, Boyko V, Reichman B, Lerner-Geva L, et al. Epidemiology and risk factors for early onset sepsis among very-low-birthweight infants. Am J Obstet Gynecol. 2009;201(1):38.e1-6.
Stoll BJ, Hansen NI, Sánchez PJ, Faix RG, Poindexter BB, Van Meurs KP, et al. Early onset neonatal sepsis: the burden of group B Streptococcal and E. coli disease continues. Pediatrics. 2011;127(5):817–26.
Palais R, Wittwer CT. Mathematical algorithms for high-resolution DNA melting analysis. Meth Enzymol. 2009;454:323–43.
Chevyrev I, Kormilitzin A. A primer on the signature method in machine learning; 2016. arXiv preprint arXiv:1603.03788 .
Morrill J, Fermanian A, Kidger P, Lyons T. A generalised signature method for multivariate time series feature extraction; 2020. arXiv preprint arXiv:2006.00873 .
Pedregosa F, Varoquaux G, Gramfort A. Scikit-learn: Machine learning in Python. … of machine Learning …. 2011.
Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, Schweer T, Peplies J, Ludwig W, Glöckner FO. The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014;42(D1):D643–8.
Austin PC, Steyerberg EW. Interpreting the concordance statistic of a logistic regression model: relation to the variance and odds ratio of a continuous explanatory variable. BMC Med Res Methodol. 2012;12:1–8.
Yang S, Ramachandran P, Rothman R, Hsieh Y-H, Hardick A, Won H, et al. Rapid identification of biothreat and other clinically relevant bacterial species by use of universal PCR coupled with high-resolution melting analysis. J Clin Microbiol. 2009;47(7):2252–5.
Athamanolap P, Hsieh K. Integrated bacterial identification and antimicrobial susceptibility testing for Polymicrobial infections using digital PCR and digital high-resolution melt in a microfluidic …. 2018 40th Annual …. 2018.
OrKeefe CM, Wang T-H lJeffr. Digital high-resolution melt platform for rapid and parallelized molecule-by-molecule genetic profiling. In: Annual international conference on IEEE Engineering in Medicine and Biology Society; 2018. pp. 5342–5.
Bagnall A, Lines J, Bostrom A, Large J, Keogh E. The great time series classification bake off: a review and experimental evaluation of recent algorithmic advances. Data Min Knowl Discov. 2017;31(3):606–60.
Kittler J, Hatef M, Duin RPW, Matas J. On combining classifiers. IEEE Trans Pattern Anal Mach Intell. 1998;20(3):226–39.
Download references
Acknowledgements
Not applicable.
This work was supported by a grant from the NIAID, Award Number R01AI134982 and a Burroughs Wellcome Fund Career Award at the Scientific Interface to S.I.F., Award Number 1012027. Fellowship support was provided to A.C.O. by an NSF GRFP award under Grant Number DGE-1650112. Support for A.B. was provided by a National Library of Medicine Grant Number 2T15LM011271-11.
Author information
Authors and affiliations.
Division of Biomedical Informatics, University of California San Diego, La Jolla, CA, 92093, USA
Aaron Boussina
Department of Nanoengineering, University of California San Diego, La Jolla, CA, 92093, USA
Lennart Langouche & Augustine C. Obirieze
Department of Bioengineering, University of California San Diego, La Jolla, CA, 92093, USA
Mridu Sinha, Hannah Mack, William Leineweber, April Aralar & Stephanie I. Fraley
Department of Pathology, University of California San Diego, La Jolla, CA, 92093, USA
David T. Pride
Department of Bioengineering, Stanford University, Stanford, CA, 94305, USA
Todd P. Coleman
You can also search for this author in PubMed Google Scholar
Contributions
S.I.F., T.P.C., M.S., A.B., L.L., and A.C.O designed the study. D.T.P. isolated and provided organisms and expertise in infectious disease. H.M., W.L., and A.A. conducted wet-lab experiments to generate the melt curve database. A.B., L.L., A.C.O. and M.S. developed and tested the algorithms. All authors read and approved the manuscript.
Corresponding authors
Correspondence to Todd P. Coleman or Stephanie I. Fraley .
Ethics declarations
Ethics approval and consent to participate, consent for publication, competing interests.
S.I.F. is a co-founder, director, and scientific advisor of MelioLabs, Inc. and has an equity interest in the company. T.P.C. is a director of MelioLabs, Inc. and has an equity interest in the company. M.S. is a co-founder and CEO of MelioLabs and has equity interest in the company. D.T.P. is an advisor of MelioLabs and has equity interest in the company. NIAID award number R01AI134982 has been identified for conflict-of-interest management based on the overall scope of the project and its potential benefit to MelioLabs, Inc.; however, the research findings included in this particular publication may not necessarily relate to the interests of MelioLabs, Inc. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict-of-interest policies. The remaining authors have no conflict-of-interest to declare.
Additional information
Publisher's note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1..
Supplementary Figures S1–S6 and Supplementary Tables S1–S2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
Boussina, A., Langouche, L., Obirieze, A.C. et al. Machine learning based DNA melt curve profiling enables automated novel genotype detection. BMC Bioinformatics 25 , 185 (2024). https://doi.org/10.1186/s12859-024-05747-0
Download citation
Received : 15 May 2023
Accepted : 14 March 2024
Published : 10 May 2024
DOI : https://doi.org/10.1186/s12859-024-05747-0
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Machine learning
- Pathogen identification
- Novelty detection
- Emerging pathogens
BMC Bioinformatics
ISSN: 1471-2105
- General enquiries: [email protected]
Featured Topics
Featured series.
A series of random questions answered by Harvard experts.
Explore the Gazette
Read the latest.

Cancer risk, wine preference, and your genes

Excited about new diet drug? This procedure seems better choice.

How friends helped fuel the rise of a relentless enemy

Christina Warinner explores “The Milk Paradox.”
Kris Snibbe/Harvard Staff Photographer
Got milk? Does it give you problems?
Biomolecular archaeologist looks at why most of world’s population has trouble digesting beverage that helped shape civilization
Harvard Staff Writer
Milk is something of a head-scratcher for Christina Warinner .
It is rich in protein, vitamins, and minerals and has served as an important food source since prehistoric times. For much of human history, milk has been consumed in various parts of the globe and has helped shape civilization, said Warriner, an anthropologist specializing in biomolecular archaeology.
But the genetic and nutritional story is complex for most of the world’s population and that has puzzled Warriner.
“We produce nearly 700 million tons of milk each year,” said Warinner, the Sally Starling Seaver Associate Professor at Radcliffe, during a Wednesday webinar, “The Milk Paradox.” “And yet we know that most of the world’s population has a lot of difficulty digesting fresh milk. So how do we get to the point where we have this food, which is spread globally and is consumed in so many different places and on every continent in various contexts, and yet it’s very difficult for us to digest?”
When humans are infants, they produce an enzyme called lactase that helps digest lactose, a sugar found in milk, but when they become adults, they stop producing it, which leads to lactose intolerance, a condition present in 65 percent of the adult human population around the world.
“When you’re an adult, you don’t produce lactase anymore, and the lactose will pass undigested into your large intestine, which is full of trillions of bacteria,” said Warinner. “They are more than happy to help you digest that lactose. The problem is in the process, because they will produce about eight liters of hydrogen gas for every quart or liter of milk that you consume.”
In her talk, Warinner, who is also the John L. Loeb Associate Professor of the Social Sciences, spoke about her interdisciplinary approach, which includes archeology, anthropology, and ethnography, to reconstruct the prehistory of milk, the origins of dairying, and its spread throughout the world.
The story of dairying took place over thousands of years from its origins in the region that encompasses West Asia, the Balkans, and North Africa to its migration to Europe and then around the world.
Scientists believed for decades that early Neolithic farmers developed a genetic mutation that allowed them to produce lactase during adulthood to properly digest milk. This change proved beneficial as they migrated to Europe, which helped them expand over the continent and replace most of the previous hunter gatherers, said Warinner.
Today lactase persistence is common in people of European ancestry as well as some African, Middle Eastern, and Southern Asian groups.
New scientific developments, including ancient DNA analysis and genome sequencing, found that there was no lactase persistence among early farmers during the Neolithic era, and raised questions about the moment when this genetic mutation took place, said Warinner.
“So this opens up a huge question, because medically we explain lactose tolerance on the basis of these mutations or adaptations,” said Warinner. “And yet for 4,000 years, people are dairying; they developed this whole food purposefully, and then they had no genetic basis to digest it. How does this work?”
It is a question that Warinner has tried to solve in her research. “I started to wonder if there might be alternative ways of adapting to a dairy-based diet that isn’t based on your own genome, but might be adapting through the use of microbes, whether that might be, for example, culinary microbes, and through fermentation, or by adapting your own gut microbiome to be able to facilitate and improve digestion.”
Warinner’s research took her to Mongolia, a country with a long history of dairying whose economy is still centered around it, and where local herders milk more species of livestock than anywhere else in the world.
Warinner’s five-year research effort included working with archeologists in Mongolia to reconstruct genomes of ancient Mongolians to see whether they showed signs of lactase persistence.
They found low lactase persistence, which is still the norm. They also studied local cattle and yak herders to analyze their gut microbiomes and compared them with those of residents in Ulaanbaatar, the capital city.
What they found was riveting, said Warinner. Their research was able to trace the history of dairying in Mongolia, which goes back 3,000 years. The analysis of hardened tooth plaque from human skeletons found in burial mounds revealed traces of milk proteins from cows, yaks, goats, and sheep.
When they analyzed lactose intolerance among herders and urban residents in Mongolia, they found that herders showed very few symptoms of lactose intolerance and low hydrogen presence. They also found that the herders’ gut microbiome showed a high volume of lactic acid bacteria, or probiotics, which may help digestion, as well as bifidobacteria, or healthy bacteria that are especially abundant in young infants and help metabolize lactose without producing any hydrogen, said Warinner.
During her talk, Warinner highlighted the way in which archaeology can help inform present-day issues or challenges, such as finding out the many ways in which humans developed dairy products and adapted to consuming milk and dairy products.
“There’s one trajectory, which is to alter the human genome, and we’ve seen this in a number of populations around the world, but an alternative pathway seems to alter the microbes that we interact with both through food and through our own gut microbiome,” said Warinner.
As for the questions guiding her next research, Warinner said, “What change caused the genetic adaptations that occurred in some populations? We don’t know what the trigger was for it, or why in Mongolia, even when it’s introduced, it was never selected for. Those are open questions that we hope to resolve.”
Share this article
You might like.
Biologist separates reality of science from the claims of profiling firms

Study finds minimally invasive treatment more cost-effective over time, brings greater weight loss

Economists imagine an alternate universe where the opioid crisis peaked in ’06, and then explain why it didn’t
Epic science inside a cubic millimeter of brain
Researchers publish largest-ever dataset of neural connections
How far has COVID set back students?
An economist, a policy expert, and a teacher explain why learning losses are worse than many parents realize
Skip to main content
- Skip to main menu
- Skip to user menu
Filter News
- All (812,422)
- Topic (770,245)
- Hotbed/Location (740,056)
- Career Advice (3,908)
- Insights (185)
- Webinars (9)
- Podcasts (46)
Integrated DNA Technologies and Molecular Health Ink Commercial Partnership
Published: May 15, 2024
Strategic partnership enables companies to combine technology offerings and streamline NGS workflows
CORALVILLE, Iowa & HEIDELBERG, Germany--( BUSINESS WIRE )-- Genomics solutions provider Integrated DNA Technologies, Inc. (IDT) and Bio-IT company Molecular Health have entered into a global multi-year agreement, starting in the U.S., to integrate their next generation sequencing (NGS) capabilities. The partnership pairs IDT’s Archer NGS research assay platform with Molecular Health’s variant annotation and reporting software to equip molecular researchers with tertiary analysis for their NGS data, maximizing lab efficiency and streamlining genomics data workflows to accelerate cancer discoveries. The collaboration also extends access to research customers in the genomic profiling space through IDT’s nationwide footprint.
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20240515774721/en/

IDT and Molecular Health's strategic partnership enables the companies to combine technology offerings and streamline NGS workflows to accelerate cancer discoveries. (Graphic: Business Wire)
“NGS remains a critical research tool used to understand the biology and pathology of cancer,” said Steve Wowk, Vice President/General Manager, Gene Reading Business Unit at IDT. “As the pace of innovation drives the cancer research community's understanding of how biomarkers are associated with the onset, progression and treatment of disease, their research now requires more complete tools inclusive of high-performance chemistries along with the ability to manage and annotate an ever-expanding biomarker knowledgebase. Our partnership with Molecular Health reflects another step toward our goal of delivering on our customers’ needs and solving their problems by equipping them with a more efficient path to advance genomic discoveries through NGS in support of the fight against cancer.”
The rapid discovery of variants that impacted the scientific field over the past five years has generated increased market demand for large targeted NGS panels, and scientists are now focused on harnessing the volume of information that these assays provide. Since the acquisition of Archer NGS research assays in 2022, IDT has been innovating on the Archer NGS platform, as reflected by this strategic partnership and three new cancer-focused product releases in 2023, including the launch of new automation-friendly liquid reagents, and RNA- and DNA-based research assays aimed at comprehensive profiling inclusive of complex genomic signatures. This continued expansion of IDT’s NGS tools portfolio has been critical for researchers looking to advance cancer discoveries.
“Cancer and other diseases can be caused by inherited or acquired genetic conditions. Understanding the consequences of mutations requires precise data generation and analysis,” said Friedrich von Bohlen, PhD, CEO of Molecular Health. “The combination of IDT's and Molecular Health’s NGS products and expertise enables high-quality, end-to-end integration plus a deep understanding of sequencing data. Our proprietary Dataome knowledge base allows clients to go beyond the generation of genomic data. It provides contextualization of variants to unlock the value of the data for researchers in the growing field of molecular profiling and discovery.”
To learn more about the partnership, or request a quote, visit https://www.idtdna.com/pages/landing/archer-request-a-consultation .
For more than 35 years, Integrated DNA Technologies, Inc. (IDT) has empowered genomics laboratories with an oligonucleotide manufacturing process unlike anyone else in the industry, with the most advanced synthesis, modification, purification, and quality control capabilities available. Since its founding in 1987, IDT has progressed from a leading oligo manufacturer to a genomics solutions provider supporting key application areas such as next generation sequencing, CRISPR genome editing, synthetic biology, digital PCR, and RNA interference. IDT manufactures products used by scientists researching many forms of cancer and most inherited and infectious diseases.
Seeking to fulfill its mission of accelerating the pace of genomics, IDT acquired Archer™ NGS Research Assays in December 2022. When combined with its existing solutions, the expanded portfolio helps realize the shared vision of enabling researchers to rapidly move from the lab to life-changing advances.
IDT’s infrastructure supports customers around the globe with manufacturing headquarters situated in Coralville, Iowa, USA, and additional manufacturing sites in San Diego, California, USA; Boulder, Colorado, USA; Research Triangle Park, North Carolina, USA; Ann Arbor, Michigan, USA; Leuven, Belgium; and Singapore.
IDT is proud to be part of Danaher. Danaher’s science and technology leadership puts IDT’s solutions at the forefront of the industry, so they can reach more people. Being part of Danaher means we can offer unparalleled breadth and depth of expertise and solutions to our customers.
Together with Danaher’s other businesses across biotechnology, diagnostics, and life sciences, we unlock the transformative potential of cutting-edge science and technology to improve billions of lives every day.
For more information about IDT, visit www.idtdna.com and follow the company on LinkedIn , X , Facebook , YouTube , and Instagram .
Disclaimer: RUO — For research use only. Not for use in diagnostic procedures . Unless otherwise agreed to in writing, IDT does not intend these products to be used in clinical applications and does not warrant their fitness or suitability for any clinical diagnostic use.
About Molecular Health
Molecular Health is a leading provider of data-driven software technology and solutions. For more than 10 years, Molecular Health has been dedicated to building software solutions that leverage a unique symbiosis of human expertise, state-of-the-art data curation, and deep analytics. The company's mission is to transform the world's biomedical knowledge into actionable insights. In the partnership with IDT, Molecular Health offers leading-edge software for analyzing genetic and molecular data.
Outside of this partnership, Molecular Health offers Pharma AI solutions for indication and biomarker finding, target identification, trial design and endpoint optimization, and safety prediction based on the Dataome technology. Molecular Health's proprietary Dataome technology integrates and contextualizes biomedical, molecular and drug-related data to provide a profound understanding of the etiology of health and disease conditions. For more information about Molecular Health, please visit www.molecularhealth.com .
View source version on businesswire.com: https://www.businesswire.com/news/home/20240515774721/en/
Kristina Sarenas Director of PR 800-328-2661 (USA & Canada) +1 319-626-8400 (outside USA) [email protected]
Source: Integrated DNA Technologies, Inc.
Smart Multimedia Gallery

View this news release and multimedia online at: http://www.businesswire.com/news/home/20240515774721/en
Back to news

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Portland Press Opt2Pay
Developments in forensic DNA analysis
Penelope r. haddrill.
Centre for Forensic Science, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, U.K.
The analysis of DNA from biological evidence recovered in the course of criminal investigations can provide very powerful evidence when a recovered profile matches one found on a DNA database or generated from a suspect. However, when no profile match is found, when the amount of DNA in a sample is too low, or the DNA too degraded to be analysed, traditional STR profiling may be of limited value. The rapidly expanding field of forensic genetics has introduced various novel methodologies that enable the analysis of challenging forensic samples, and that can generate intelligence about the donor of a biological sample. This article reviews some of the most important recent advances in the field, including the application of massively parallel sequencing to the analysis of STRs and other marker types, advancements in DNA mixture interpretation, particularly the use of probabilistic genotyping methods, the profiling of different RNA types for the identification of body fluids, the interrogation of SNP markers for predicting forensically relevant phenotypes, epigenetics and the analysis of DNA methylation to determine tissue type and estimate age, and the emerging field of forensic genetic genealogy. A key challenge will be for researchers to consider carefully how these innovations can be implemented into forensic practice to ensure their potential benefits are maximised.
Introduction
Since its first use in a criminal case in 1987, the analysis of DNA from biological evidence has revolutionised forensic investigations. The intervening three decades have seen significant advancements in terms of the discrimination power, speed, and sensitivity of DNA profiling methods, as well as the ability to type increasingly challenging samples [ 1–3 ]. The establishment of databases of offender and crime scene profiles, and of population allele frequencies, have permitted the identification of suspects from crime scene samples and the development of statistical frameworks for evaluating DNA evidence [ 1 , 2 ]. Recent years have seen the expansion of the number of loci included in short tandem repeat (STR) typing kits and standardisation of core loci across jurisdictions, allowing for greater cross-border sharing of DNA profiling data [ 4 , 5 ]. When a recovered profile matches one found on a DNA database, or generated from a suspect, DNA evidence can thus be extremely powerful.
However, when no profile match is found, when the amount of DNA in a sample is too low, or the DNA too degraded to be analysed, traditional STR profiling may be of limited value. The introduction of novel techniques and technologies into the criminal justice system is slow, but new methodologies are being developed that enable the analysis of these challenging samples, and that can generate intelligence about the donor of a biological sample [ 3 , 5 ]. The last few years have seen a rapid expansion in the field of forensic genetics (and now forensic genomics), demonstrated by growing numbers of publications in the field over the last two decades ( Figure 1 ). An exhaustive review of this whole field is beyond the scope of a single article, and so this review seeks to provide an overview of some of the most important recent advances in the forensic analysis of DNA.

Massively parallel sequencing
Massively parallel sequencing (MPS) technologies, frequently referred to as next-generation sequencing (NGS) technologies, have revolutionised the biological sciences by their ability to generate millions of sequencing reads in a single run. Despite only relatively recently being adopted in the forensic field, the use of MPS for forensic applications has expanded rapidly in the last few years [ 6 , 7 ]. Whilst MPS has permitted high-throughput sequencing of the whole genomes of a huge variety of organisms, forensic applications have used a more targeted approach, including an initial PCR-amplification of a set of target markers prior to MPS of the resulting amplicons [ 8 ]. There are two main technology platforms used for forensic applications of MPS, the Illumina sequencing-by-synthesis method, and Thermo Fisher's semiconductor-based Ion Torrent sequencing [ 9 ]. A variety of kits are available for use on each platform, targeting different forensically relevant markers, and a growing number of studies have validated and/or evaluated these kits for forensic use (reviewed in [ 9–11 ]). For example, the Precision ID Globalfiler™ NGS STR Panel and Precision ID Identity/Ancestry Panels use the Ion S5 system to sequence STR and single nucleotide polymorphism (SNP) markers [ 12 , 13 ], and Promega's PowerSeq® system sequences autosomal and Y-STRs on the Illumina MiSeq [ 14 ]. The Verogen ForenSeq™ DNA Signature Prep Kit runs on the MiSeq FGx™ Forensic Genomics System, which uses Illumina technology to sequence a combination of autosomal STRs, Y-/X-STRs, and identity SNPs, and can be expanded to include phenotype- and ancestry-informative SNPs [ 15 , 16 ]. Kits have also been developed to target part or all of the mitochondrial genome [ 17–19 ].
This ability to target large numbers of different marker types into a single assay is one of the key advantages of MPS methods, increasing discrimination power and of particular benefit when analysing the often-limited DNA in forensic samples [ 20 ]. Another major advantage of MPS technology is that it detects nucleotide sequence variation in the targeted markers, including variants in STR repeat regions and flanking sequences [ 21 ]. This permits discrimination of alleles that would be indistinguishable using capillary electrophoresis length-based typing ( Figure 2 ), a feature that also has advantages for interpretation of complex mixed profiles [ 22 ]. MPS also improves results for low level and degraded DNA samples, as a result of the shorter amplicons compared with standard STR profiling [ 23 , 24 ].

All three of these alleles would be classified as 14 alleles on the basis of their length but determining their nucleotide sequence allows them to be discriminated.
There are a number of barriers to widespread adoption of MPS for forensic applications, including variable performance of some markers in terms of coverage and locus imbalance [ 11 ], and susceptibility to PCR inhibitors when compared with standard STR typing kits [ 25 , 26 ], but these are rapidly being overcome. Development of a standardised nomenclature system, which captures sequence variation in MPS-generated STR alleles whilst maintaining compatibility with existing CE-based STR data in national DNA databases, is a particular challenge, but recommendations are now in place to address this [ 27–30 ]. The availability of frequency data for alleles detected using MPS is also increasing with the publication of datasets from populations worldwide [ 31–35 ]. The costs of MPS are decreasing all the time and with the development of bioinformatics tools to analyse the large volume of complex data produced, implementation of MPS technologies into forensic workflows is becoming realistic. Although MPS methods have not yet been widely implemented in casework, with wider applications in other areas of forensic analysis (e.g. RNA sequencing, epigenetics, forensic DNA phenotyping; see below) these technologies are likely to become indispensable tools for the forensic community.
DNA mixture interpretation
Interpretation of DNA profiles containing contributions from multiple donors is much more complicated than single source profiles ( Figure 3 ), not only because of the potential number of alleles present in the profile, but also because such profiles are often low-level with complicating features such as allele drop-out/drop-in and heterozygous imbalance [ 36 ]. The increasing sensitivity of STR profiling techniques means that the recovery of mixed DNA profiles has become more common, not only from samples where mixtures might be expected (e.g. sexual offence samples), but also from low quality/quantity samples recovered from handled items [ 37 ]. Such samples often produce complex mixtures, with large numbers of contributors and no individual who can be assumed to be present in the mixture [ 38 ].

Across a whole profile, interpretation of the varying number of peaks and peak heights can become very complex, even with a small number of contributors.
This increasing complexity of mixed profiles has called for increasingly complex methods of mixture interpretation, and there has been a move away from relatively simple methods that ranged from determining whether an individual could be excluded as a potential contributor to a mixture, to the use of likelihood ratio methods that estimated the most likely genotype combinations of contributors to a mixture, the more complex of which used some of the information contained within profile peak heights [ 39 , 40 ]. This has led to the development of mixture interpretation methods using probabilistic frameworks, incorporating probabilities of allele drop-out and drop-in, modelled from validation and empirical data [ 41 ]. These probabilistic genotyping methods are broadly categorised as semi-continuous, which do not utilise peak height information or model artefacts such as stutter, and continuous, which do [ 42 ]. The complexity of the statistical calculations involved in these methods is such that specialised software is required to carry out these analyses, and there are a variety of programmes now available for this purpose (summarised in [ 5 ]). The ability of these programmes to analyse mixtures previously considered too complicated for interpretation has seen rapid uptake by forensic laboratories, and publication of studies reporting the developmental and internal validation of different probabilistic genotyping software packages, as well as guidelines for their use by a number of regulating bodies [ 43–48 ].
The software packages that implement probabilistic genotyping methods are highly complex, and developers have urged forensic laboratories to ensure their analysts have a good understanding of the concepts underlying the methods and that they remain involved in the interpretation of profiles and critical evaluation of the mixture analysis [ 36 , 49 ]. Concerns have been raised over variation in the output of probabilistic genotyping methods, some due to subjective decisions made by the user, some due to variability inherent in the methods [ 50 , 51 ]. Some countries have seen extensive debates over the admissibility of probabilistic genotyping methods in court and whether methods have gained general acceptance in the community, but the widespread implementation of these methods into forensic laboratories around the world suggests they have [ 42 ]. These methods also provide significant promise for the interpretation of MPS data that can uncover greater complexity in mixed profiles by identifying sequence differences between alleles that would be indistinguishable by length ( Figure 2 ).
Body fluid identification
The ability to identify the presence of a specific body-fluid can be extremely valuable to an investigation, providing crucial information on the activities involved in an incident, particularly if it means that a DNA profile can be linked to a specific biological source. All of the presumptive/confirmatory tests currently used to identify some (but not all e.g. vaginal material, menstrual blood) body fluids have limitations, including a lack of sensitivity and specificity, and a requirement to carry out multiple tests that destroy limited samples [ 52 ]. This has led to interest in the analysis of RNA in body fluid stains, particularly given RNA can be co-extracted with DNA, allowing parallel production of a DNA profile alongside body fluid testing [ 53 ].
Identification of body fluids using RNA profiling is based on the principle that although DNA content is the same in most cell types, RNA differs depending on cell type and function. The production of RNA is therefore tissue-specific, such that each body fluid has a specific gene expression pattern. The presence of tissue-specific RNA types in a sample can therefore indicate the presence of specific body fluids [ 54 ]. Research in this area has focused on large-scale screens for differentially expressed RNAs followed by the development of PCR-based assays to target individual or small numbers of markers. Many of these assays employ reverse transcription endpoint (RT-PCR) or quantitative real-time (RT-qPCR) PCR [ 55 ], which have the benefit of compatibility with existing technologies in forensic laboratories, although increasingly studies are utilising the power of MPS for identification and analysis of tissue-specific RNAs [ 56–59 ].
Initial assays focused on identifying body fluid-specific messenger RNA (mRNA) markers, and development of multiplexes indicating the presence of single or multiple body fluid types, the latter of which is particularly useful when analysing mixed samples (reviewed in [ 55 , 60 ]). However, the susceptibility of mRNAs to degradation has limited their application to forensic samples, and mRNA assays also suffer from limitations including variation in sensitivity and specificity and interpretational challenges [ 61–64 ]. More recently, focus has been on micro RNAs (miRNAs) as alternative markers for body fluid identification [ 65 ]. Many of these regulatory RNAs, which target mRNAs for degradation or silencing, also show tissue-specific expression, and have the benefit of increased stability compared with mRNA as a result of their smaller size and incorporation into a protein complex within the cell [ 66 ]. A variety of miRNAs have been identified as potential markers for forensically relevant body fluids, and although it is unlikely any miRNAs are specific to single body fluid types, a number of assays have been developed that incorporate panels of multiple differentially expressed miRNAs that appear to identify specific body fluids [ 67–73 ]. Although miRNA-based assays suffer from some of the same challenges as mRNA-based assays, particularly in terms of interpretation, miRNAs have great potential as body fluid markers [ 67 , 68 , 70 ]. Further study to identify the best sets of miRNAs to unambiguously identify different body fluids and detailed validation of the resulting assays may give the forensic community a reliable test for the identification of body fluids [ 65 ]. Micro RNA markers also hold promise for other forensic applications, including estimating the time of deposition of body fluid stains [ 74 , 75 ] and the post-mortem interval [ 76 ].
Forensic DNA phenotyping
When standard STR profiling fails to advance an investigation because no match to a known suspect or DNA database is found, any information that can assist in identifying the donor of the sample would be very valuable. This has led to the development of tests that predict externally visible characteristics (EVCs) from DNA samples, which can provide intelligence leads to investigations, narrowing the pool of potential suspects [ 77 ]. The ability to predict an individual's appearance also has utility in missing persons cases and in disaster victim identification. More widely, forensic DNA phenotyping (FDP) is considered to encompass the prediction of EVCs, inference of bio-geographic ancestry, and the estimation of age using epigenetic markers [ 78 , 79 ].
FDP techniques have developed from many decades of research identifying SNPs that are statistically associated with particular characteristics, via genome-wide association studies [ 80 ]. From this, small sets of SNPs have been identified that can be typed in PCR multiplexes and analysed using statistical models that predict EVCs of interest with high accuracy. By far the most advanced and successful of these relate to the prediction of human pigmentation traits [ 77 ]. The genetics of physical traits is often complex, with the expression of many traits controlled by variation at a large number of genes, as well as environmental factors [ 81 ]. However, human pigmentation traits are influenced by a relatively small number of genes compared with other traits, and it is these pigmentation traits that have been the focus of FDP, principally eye and hair colour and, more recently, skin colour [ 77 ].
A number of test systems have been developed for the prediction of human pigmentation traits, including the forensically validated IrisPlex [ 82 , 83 ], HIrisPlex [ 84 , 85 ], and HirisPlex-S [ 86 , 87 ] assays, which predict broad categories of eye, hair, and skin colour by analysing 6, 24, and 41 SNPs, respectively. Inclusion of additional SNPs and improvements in prediction models means that these pigmentation traits can now be predicted with good accuracy, usually expressed using a measure known as the AUC (area under the receiver operating characteristic curve), which can take values from 0.5 (random prediction of the characteristic) to 1.0 (accurate prediction) [ 77 ]. For example, the most recent IrisPlex model for eye colour prediction gives accuracies of 0.95 for brown, 0.94 for blue, and 0.74 for intermediate (e.g. non-blue and non-brown) eye colours, reflecting the fact that intermediate eye colours are predicted with lower accuracy as the genetic variants responsible for these colours are less well understood. This resulted in an average eye colour prediction accuracy of 84%, or 93% when only blue and brown categories were included [ 85 ]. Similarly, the HIrisPlex model results in hair colour prediction accuracies of 0.92 for red, 0.85 for black, 0.81 for blond, and 0.75 for brown hair colour, giving an average hair colour prediction accuracy of 73% [ 85 ]. The accuracy of hair prediction is influenced by the phenomenon of hair darkening with age, which can lead to the prediction of a lighter hair colour than the observed phenotype, and this predominantly affects individuals who are categorised as having brown hair but predicted to be blond [ 84 ]. More recently, the accuracy of the HIrisPlex-S skin colour prediction model has been assessed using both 3 and 5 skin colour categories, resulting in AUC values of 0.97 for light, 0.96 for dark-black, and 0.83 for light skin colour categories, and 0.97 for dark-black, 0.87 for dark, 0.74 for very pale, 0.73 for intermediate, and 0.72 for pale skin colour categories [ 86 , 87 ].
The systems described above use multiplex PCR followed by multiplex single-base extension using SNaPshot chemistry, which is limited in terms of the number of SNPs that can be typed in a single multiplex. More recently, the ability of MPS technologies to analyse very large numbers of genetic markers in a single run, even at low levels of input DNA, has been exploited to develop both commercial and custom assays that predict ancestry and/or EVCs [ 88 , 89 ]. For example, researchers in the VISAGE Consortium have combined the 41 SNPs in the HIrisPlex-S system with 115 SNPs that provide information about bio-geographical ancestry to generate an assay that can be run on different MPS platforms [ 90 , 91 ]. VISAGE, the VISible Attributes through GEnomics Consortium, is an EU-funded collaborative research program established in 2017 to work towards the provision of intelligence information about an individual's appearance, age, and ancestry from DNA recovered in the course of investigations ( http://www.visage-h2020.eu ). The consortium also has a focus on the complex legal, regulatory, and ethical issues surrounding the prediction of EVCs from DNA for forensic purposes, which in many countries is not currently subject to any specific legislation [ 92 , 93 ]. VISAGE researchers and others also continue to make progress on the development of systems to predict other EVCs, including eyebrow colour [ 94 ], stature [ 95 ], skin features such as freckles and tanning [ 96 ], and further hair-related phenotypes such as head hair shape [ 97 ] and age-related hair darkening [ 98 ].
Epigenetics and DNA methylation analysis
In addition to the information encoded within the sequence of DNA bases in the genome, the DNA molecule carries an additional layer of information in the form of chemical modifications of nucleotides and chromatin-related proteins [ 99 ]. Broadly defined as epigenetic changes, these modifications alter patterns of gene expression via a variety of mechanisms and have been shown to have a role in the regulation of key cellular processes, with epigenetic errors being associated with diseases such as cancer [ 100 ]. The addition of a methyl group (-CH 3 ) to the 5′ position of cytosine residues in the human genome, primarily those found in cytosine-guanine dinucleotides (known as CpG sites), is one of the most widely studied epigenetic modifications, and observations of differential methylation patterns with age and across tissue types has led to interest in forensic applications of DNA methylation analysis [ 101 ]. Research in this area has focused on estimation of the age of the donor of a DNA sample and the identification of tissue-type for body fluids and other forensically relevant biological samples [ 102–105 ], although DNA methylation analysis has a range of other forensic applications including the discrimination of monozygotic twins [ 105 ] and the determination of smoking status [ 106 ].
DNA methylation plays a crucial role in cell differentiation, such that CpG sites are differentially methylated in different tissues [ 100 ]. This tissue-specificity has been successfully exploited for the development of methylation-based assays using a range of technologies [ 103 , 104 ]. Numerous studies have described epigenetic markers for the identification of forensically relevant tissue-types, including blood, semen, saliva, vaginal material and menstrual blood, (e.g. [ 107–114 ]), some of which pose more significant challenges than others [ 115–117 ]. A key benefit of identifying tissue type from the analysis of DNA rather than RNA is that this may provide a link between an STR profile and the corresponding tissue type, given both types of information come from the same molecule [ 79 , 103 ].
The ability to predict the chronological age of an unknown individual from a DNA sample could provide extremely useful intelligence to investigators, particularly in combination with the prediction of EVCs, many of which can vary with age [ 103 ]. A number of authors have identified CpG sites where methylation level is correlated with age and built age-prediction models targeting small numbers of sites that can be incorporated into assays to estimate age with high accuracy, using technologies such as pyrosequencing, single base extension using SNaPshot chemistry, and EpiTyper (e.g. [ 118–120 ]). Universal age-related markers would have significant benefits in terms of developing models to predict age across multiple tissues, but the highest age-prediction accuracy has been seen in tissue-specific models [ 102 , 104 , 121–123 ]. These have mainly focused on whole blood [ 124–128 ], with some studies on other tissues such as saliva and semen [ 115 , 129–132 ]. A number of MPS-based targeted methylation assays for age prediction have also now been developed, overcoming the multiplexing limitations of previous technologies and permitting analysis of multiple CpG sites in a single highly sensitive assay [ 104 , 127 , 133 , 134 ]. These age prediction assays estimate age with high accuracy, measured in terms of the mean absolute deviation (MAD) between the estimated and chronological age, with many assays providing prediction accuracies of ±3–4 years [ 79 , 103 ].
Genetic genealogy
Since the high-profile arrest in 2018 of Joseph DeAngelo as a suspect in the Golden State Killer investigation, attention has focused on the applications of genetic genealogy in a forensic context [ 135 , 136 ]. Whilst familial searching of forensic DNA databases has been effectively used to identify close (first/second degree) relatives of suspects via the detection of allele sharing in STR profiles, genealogists can identify much larger numbers of more distant relatives (third to ninth degree) by detecting stretches of DNA in the genome that are identical by descent, indicating common ancestry [ 137 ]. This is achieved by exploiting huge genetic datasets amassed by individuals taking direct to consumer (DTC) genetic tests for the purposes of genealogical research. These tests type hundreds of thousands of autosomal SNP variants, the results of which are then shared on large public platforms such as GEDmatch ( https://www.gedmatch.com/ ) that allow testers to identify potential relatives [ 138 , 139 ]. Searching of these online platforms using profiles generated from samples recovered in criminal investigations may identify relatives of the potential perpetrator, and further genealogical research may lead to the identification of a suspect whose DNA can then be recovered and compared with crime samples [ 140 ].
The size of these public databases of genetic information are such that one study estimated 60% of searches were likely to find a relative at a distance of third cousin or closer, and 15% second cousin or closer, indicating that a database covering only 2% of a target population would include a third cousin match for 99% of the population ([ 141 ], see also [ 142 ]). The vast majority of people who have taken DTC genetic tests are US citizens of European ancestry, and their over-representation in genealogical databases means the chances of finding relative matches are significantly higher for this population [ 140 , 141 ]. However, there is increasing interest in other populations, both in the uptake of DTC genetic testing by the public and the mining of this data by law enforcement. Since the arrest of DeAngelo and a number of other notable success stories, law enforcement agencies worldwide have begun to see the potential in this approach for identifying the distant relatives of suspects [ 138 ].
The approach has sparked concerns about data privacy and ethics, as a result of surreptitious law enforcement searches of public databases, although the majority of platforms that are accessible to law enforcement agencies now either offer consumers the option to opt-out if they do not want their data being included in these types of searches, or explicitly require them to opt in [ 140 , 143 ]. There are ongoing concerns about sharing and privacy of genetic data and the legality of these types of search, as well as the ethics of individuals who have not taken a genetic test being exposed to attention from investigators because a relative has [ 137 , 144–146 ]. There are also no validation studies of genealogical techniques for forensic use [ 138 , 139 ], but the techniques are used only to generate intelligence leads in investigations, often when they have been cold for many years, and any leads would always be verified using standard STR profiling [ 140 ]. With the recent acquisition of GEDmatch by forensic genomics company Verogen ( https://verogen.com/gedmatch-partners-with-genomics-firm/ ), along with the launch of a kit specifically designed for genealogical applications ( https://verogen.com/products/forenseq-kintelligence-kit/ ), it seems likely that these methods will become commonplace in investigations.
Conclusions
This review highlights some of the recent key developments in the rapidly expanding field of forensic genetics, but there are many other exciting areas of research that could not be covered here. For example, methodological developments in DNA extraction [ 147 ], direct PCR [ 148 ], and rapid/at-scene processing of samples on portable devices (reviewed in [ 5 ]), the application of MPS technologies to new marker types such as microhaplotypes [ 149 , 150 ], analysis of non-human DNA in the form of human and environmental microbiomes [ 151 , 152 ], and the use of third-generation sequencing devices in forensic DNA analysis [ 9 , 11 ] represent just some of the current and future developments in the field. It will be crucially important that researchers consider how to harness the innovations produced by this dynamic field to ensure their implementation into forensic practice.
- The analysis of DNA from biological material recovered in the course of a criminal investigation can provide very powerful evidence, however when there is no match between the recovered profile and a DNA database or suspect the evidence may be of limited value.
- The rapidly expanding field of forensic genetics research has introduced various novel methods that enable the analysis of challenging forensic samples, and that can generate intelligence about the donor of a biological sample.
- This article reviews some of the most important advances in the field, including the application of massively parallel sequencing, advancements in DNA mixture interpretation, body fluid identification using RNA profiling, forensic DNA phenotyping, epigenetics and DNA methylation analysis, and genetic genealogy.
- A key challenge will be to ensure that the benefits of these novel technologies can be maximised by implementing them into forensic practice.
Acknowledgements
The author thanks Arthur Cleal for permission to use the image in Figure 3 , and two anonymous reviewers for very helpful comments on the manuscript. No funding was received in support of this work.
Abbreviations
Competing interests.
The author declares that there are no competing interests associated with this manuscript.
Open Access
Open access for this article was enabled by the participation of University of Strathclyde in an all-inclusive Read & Publish pilot with Portland Press and the Biochemical Society under a transformative agreement with JISC.
2024 DNA Internship (Domestic)
工作地點 台灣 職別 其它 職務類型 實習 職務張貼日 2024/01/05
TSMC Summer Intern Program provides an unique experience for students to learn about the semiconductor industry and have a first-hand experience of career life in TSMC. You will not only join a series of training sessions related to your major, but also be part of the team and expected to provide solutions and create valuable impacts to our company.
For Engineering field related positions, we welcome all the talents from STEM fields (Electronics, Electrical Engineering, Physics, Material Science and Engineering, Chemistry, Chemical Engineering, Mechanical and Automation Engineering, Computer Science, Industrial Engineering) to join us!
For Non-Engineering field related positions, we welcome all the talents from various background including, Business, Supply Chain Management, Human Resources, Finance, Legal, Corporate Planning, Material Management & Risk Management major's students to join us!
Program Timeline
Application Time : Now -2024/04/30
Interview Period : Now-2024/06/28
Internship Program Time:2024/07/01 to 2024/08/30
2023 DNA Internship highlights : https://www.youtube.com/watch?v=DFliJzhGOcg
For Engineering Field Related Positions
1. Equipment Engineer (EE)
In this Equipment Engineer intern role, you will be closely learning with professional engineers to plan and execute the analysis or defect detection projects. You can also learn how to improve and enhance the efficiency of equipment by communicating with cross function engineers or vendors. Don't miss out the opportunity to gain a real competitive edge with us!
2. Process Integration Engineer (PIE)
In this PID role, you will learn to develop and sustain process technologies for flash memory and logic products. You will also have opportunities to work with a team which may include device, integration, yield, lithography, etch and thin films or external suppliers to drive leading-edge integrated module development, control and improvements. Sing up and build your career networks and professional experiences with TSMC now!
3. Process Engineer (PE)
During this internship, you will have unique chances to experience how to work with a team which may include device, integration, yield, module, manufacturing and external suppliers to drive leading-edge integrated module development, control and improvements. You will also learn to process stability/manufacturability improvement for yield and reliability qualification. Come and join one of the world’s largest semiconductor manufacturing companies!
4. Intelligent Manufacturing Engineer (MFG)
During this internship, you will be overseeing the daily operations of IC foundry to ensure that all profiling operations, workflow, and customer reports are consistent with agreed upon service operations. You will also learn to analyze production related data and provide dynamical strategies to achieve organization production goals & KPI. Discover TSMC and thrive with us today!
5. Semiconductor Process R&D
Do you want to make a difference to the world? If yes, join our exceptional R&D team today! You will experience and learn with our professional R&D colleagues about how to conduct exploratory research in advanced technologies, evaluate a variety of advanced materials, equipment/ tools, process/ Recipe tuning, and handle advanced device, process integration. Come and pursue the bright future with TSMC!
6. IC Design
In this IC Design intern role, you will learn how to communicate between TSMC customers and our process technology development team. You will also learn the basic way to drive process-design optimization to ensure technology leadership, reduces customer's barrier for adopting TSMC technology, and enables TSMC business through design differentiation. Don't miss out the opportunity to gain a real competitive edge with us!
7. Intelligent Manufacturing Engineer
During this internship, you will have unique chances to experience how to run the production and supervises the daily operations of IC foundry to ensure smooth workflow. You will learn to address field-related issues alongside your peers and with the support of leadership. Don't miss out the chance to work in one of the world’s largest semiconductor manufacturing companies!
8. Information Technology
If you're looking for an exciting, challenging, and rewarding career, become a IT Intern at TSMC today! In this role, you will learn how TSMC enhance competitive advantage through cost-efficient, robust, and scalable IT systems and solutions, including improving productivity, accelerating innovation, empowering customer service, enabling decision-making. Don't hesitate to grow beyond your capacity with us!
9. Quality & Reliability
As a Quality & Reliability Intern, you will learn the basic knowledge of raw material supplier quality management and operation of chemical analysis laboratory and how TSMC Quality and Reliability team conduct risk assessment of analysis result to ensure production quality. Join us and gain meaningful working experiences and essential skills for your bright future!
For Non-Engineering Field Related Positions
1. Human Resources
If you're eager to gain an insight in the HR field, don't hesitate to join our professional HR team to learn how to deal with daily HR administrative across the organization. Meanwhile, you will also have chances to partner with different recruiters to better understand company's needs to optimize recruiting strategy. Don't miss out the opportunity to gain a real competitive edge with us!
2. Accounting
In accounting department, we will prepare students to become highly talented in Finance / Accounting background to explore a career in corporate accounting. Interns will be assigned to specific department based on the candidate’s background, skill set and interests. The assignment will be project-based, relevant to managerial accounting, tax planning or IFRS related, allow you to take responsibility for a major task. And you will experience a sense of personal accomplishment as well as TSMC’s culture through project assignment and structured activities.
As a finance intern, you will have exciting chances to apply your classroom knowledge in a global business practice. By joining us, you will participate in various corporate finance projects by engaging with different functions of the Finance Division, including Financial Planning, Investment Management, Foreign Exchange Management, etc. You will also learn fundamental concepts of corporate finance and essential skills that will prepare you to become a professional financier. Discover TSMC and thrive with us today!
In this legal intern role, you will have room to grow your strengths and show us what you can do. During the internship, you will enhance basic knowledge of how to facilitate compliance with domestic and international regulations. You will also be working closely with our legal advisors to learn some basic practices by dealing with various corporate legal cases at TSMC. Join us to achieve your career goal today!
5. Corporate Planning
Are you excited to roll-up your sleeve and show us what you learned from school? TSMC Corporate Planning Organization will help you learn the fundamental methodologies of how to design flexible planning to efficiently coordinate the needs between factories and customers. Also, you will have chances to collaborate with cross-function members to manage logistic service related projects. Not only can you boost experiences during this internship, but learn practical skills and knowledge all at the same time!
6. Material Management & Risk Management
Are you interested to work in a large scale business environment? If yes, join our MM&RM internship to gain real-world experiences in learning demand forecast and supply planning. You will also coordinate with cross-function members to conduct projects related to logistics/warehouse system/process enhancement. Sing up and build your career networks and professional experiences with TSMC now!
Monthly Salary Range: from $33,000
For Engineering Field Related Positions:
(1) Bachelor's degree or above in Electronics, Electrical Engineering, Mechanical and Automation Engineering related fields.
(2) Have basic mechanical related knowledge.
(3) Good problem-solving skills, communication ability, team spirit, active learning attitude.
(4) Hands-on participation and a strong sense of ownership.
(1) Bachelor's degree or above in an engineering and scientific field such as Materials Science, Engineering, Electrical Engineering, Chemical Engineering, Physics, Chemistry.
(2) Exhibit good and open communication skills, be able to work within cross-functional teams, including internal and external partners.
(3) Hands-on participation and a strong sense of ownership.
(1) Bachelor's degree or above in Materials Science, Electrical Engineering, Chemical Engineering, Physics, Chemistry or a related engineer discipline.
(2) Exhibit good and open communication skills, be able to work within cross-functional teams.
(3) Strong technical problem-solving and analytical skills.
(1) Bachelor's degree or above in Industrial Engineering, Manufacturing, Production Engineering, Industrial Management, Information System, Mechanical and Automation Engineering, Civil, Applied Mathematics and statistics or related field.
(2) Must be an enthusiastic and dedicated individual with the capability to motivate and lead using a team approach.
(3) Having the semiconductor processes knowledge is a plus.
(1) Master's degree or above in chemistry, materials science, physics, electrical engineering or related science and engineering fields
(2) Intermediate or above level of communication skills in both English and Mandarin
(3) Logical thinking, teamwork spirit, and good communication skill
(4) Positive attitude to confront difficulties and challenges
(5) Ability to work and operate effectively in a fast-paced environment
(1) Master's degree or above in EE/CS Background
(1) Master's degree or above in Engineering-Related and Industrial Engineering Background
(1) Bachelor's degree or above and major in Computer Science, Information engineering, Industry Engineering, Statistics or Mathematic related fields, similar technical field of study or equivalent practical experience.
(2) Experience in JavaScript, Java, C#, Python, Kotlin, Rust or other relevant coding languages is a plus
(3) Knowledge about IC process and logistic flow shall be better to have
(4) Intermediate to Excellent level of communication skills in both English and Mandarin
(5) Logical thinking, teamwork spirit, and good communication skill
(6) Positive attitude to confront difficulties and challenges
(7) Ability to work and operate effectively in a fast-paced environment
(2) Statistics or machine-learning backgrounds for data analysis and statistics development & application related roles is a plus.
(3) Intermediate or above level of communication skills in both English and Mandarin
(4) Logical thinking, teamwork mindset, and good communication skill
(5) Positive attitude to confront difficulties and challenges
(6) Ability to work and operate effectively in a fast-paced environment
(1) Bachelor's degree or above in human resource, psychology, social sciences, and management-related majors
(2) Excellent interpersonal and communication skills
(4) Logical thinking, teamwork spirit, and good communication skill
(1) Working toward an Accounting or Finance Degree and have completed courses within major
(2) Proficiency in MS Excel and MS PowerPoint
(3) Strong written and verbal communication skills in English and Mandarin
(1) Bachelor's degree or above in business and finance-related majors
(2) Strong financial knowledge and analytical skills
(1) 1L of top law schools with good academic performances
(2) Completion of fundamental law courses or equivalent work experience is a plus
(1) Bachelor's degree or above in IE or Business
(2) Cross function/organization project handling experience is a plus
(5) Positive attitude to confront difficulties and challenges
(1) Master's degree or above in engineering/business related fields
(2) Cross function/organization project handling experience is a plus
IMAGES
VIDEO
COMMENTS
Salting-Out Method. Introduced by Miller et al 55 in 1988, this method is a nontoxic DNA extraction method. Procedure: Sample is added to 3 mL of lysis buffer, SDS, and proteinase K, and incubated at 55 to 65°C overnight. Next, 6 mL of saturated NaCl is added and centrifuged at 2,500 rpm for 15 minutes.
Advances in genetics, genomics and molecular biology are improving existing forensic approaches and providing new ones. The authors discuss improvements in DNA profiling, the growing field of ...
† 1984: Alec Jeffrey introduced DNA fingerprinting in the field of forensic genetics, and proved thatsomeregions in the DNA have repetitive sequences, which vary among individuals. Due to this discovery, first forensic case was solved using DNA analysis.8 DNA Structure and Genome DNA was first described by Watson and Crick in 1953, as
DNA Profiling in Forensic Science Bukyya et al. 139 Silica Column-Based DN AE x t r a c t i o nM e t h o d In this method, 1% SDS, lysis buffer (3 mL of 0.2 M tris and
According to The American Heritage Medical Dictionary, DNA profiling is "the identification and documentation of the structure of certain regions of a given DNA molecule, used to determine the source of a DNA sample, to determine a child's paternity, to diagnose genetic disorders, or to incriminate or exonerate suspects of a crime []."DNA profiling (also named DNA typing, DNA ...
DNA profiling, as it has been known since 1994, has been used in the criminal justice system since the late 1980s, and was originally termed "DNA fingerprinting". The DNA in every human is ...
The origin of DNA fingerprinting, or more correctly DNA typing or DNA profiling is generally attributed [] to the work of Alec Jeffreys, an English geneticist who, in 1985, found and isolated regions of DNA containing DNA sequences that repeated themselves many times throughout the DNA (and are therefore relatively easy to isolate) and that are characterized by high polymorphism, that is they ...
Handbook of DNA Profiling Reference work ... DNA Fingerprinting Unit, State Forensic Science Laboratory, Sagar, India ... India. Besides teaching and research, he is actively involved in conducting training programs to various beneficiaries of forensic science. Before joining academics, he served as a forensic DNA expert at Madhya Pradesh ...
Introduction. The first use of DNA fingerprinting was to resolve an immigration dispute in 1985. Citation 4 A process of DNA fingerprinting had been published the year earlier in Nature, coauthored by Alec Jeffreys.This led to a local police force engaging Jeffreys (now Professor Sir Alec Jeffreys) to assist with an unsolved sexual assault and murder of two young girls in Leicestershire, England.
DNA Fingerprinting. E. Giardina, in Brenner's Encyclopedia of Genetics (Second Edition), 2013 Introduction. DNA fingerprinting (also called DNA profiling or forensic genetics) is a technique employed by forensic scientists to assist in the identification of individuals or samples by their respective DNA profiles. Although more than 99.1% of the genome is the same throughout the human ...
The role of DNA in forensic science: A comprehensive review. August 2023. International Journal of Science and Research Archive 9 (2):814-829. DOI: 10.30574/ijsra.2023.9.2.0624. Authors: Salem K ...
DNA profiling is a forensic technique in criminal investigations, comparing criminal suspects' profiles to DNA evidence so as to assess the likelihood of their involvement in the crime. [1] [2] It is also used in paternity testing, [3] to establish immigration eligibility, [4] and in genealogical and medical research.
DNA profiling is used to identify an individual from a sample of DNA by looking at unique patterns in the DNA sequence. On average, about 99.9% of the DNA between two humans is the same. The remaining 0.1% might not sound like much - but that's around 3 million bases that can differ between two people. DNA profiling looks at these ...
DNA profiling. DNA profiling is the process where a specific DNA pattern, called a profile, is obtained from a person or sample of bodily tissue. Even though we are all unique, most of our DNA is actually identical to other people's DNA. However, specific regions vary highly between people. These regions are called polymorphic.
DNA. Deoxyribonucleic acid is a macromolecule containing the genetic instructions for the replication and function of all known living organisms (excepting a small number of viruses). Specific segments known as genes carry this genetic information and encode for proteins, for example, are known as genes.
Use your DNA fingerprinting technology to prove he killed both girls, they asked him. So Jeffreys set up his tests, using - in this case - a version of DNA fingerprinting called DNA profiling.
DNA fingerprinting, one of the great discoveries of the late 20th century, has revolutionized forensic investigations. This review briefly recapitulates 30 years of progress in forensic DNA analysis which helps to convict criminals, exonerate the wrongly accused, and identify victims of crime, disasters, and war. Current standard methods based on short tandem repeats (STRs) as well as lineage ...
It does this using a repeating process that takes about five minutes. First, you add a heat-stable DNA polymerase — a special enzyme that binds to the DNA and allows it to replicate. Next, heat the DNA sample to 200 degrees F (93 degrees C) to separate the threads. Then let the cool before reheating it.
25 Altmetric. Metrics. We developed a two-pronged strategy to functionally probe the enormous repertoire of noncoding DNA within genomes. Our approach markedly improved signal-to-noise ratio and ...
Since HRM-based DNA profiling is an inexpensive and rapid technique, our results add support for the feasibility of its use in surveillance applications. Surveillance for genetic variation of microbial pathogens, both within and among species, plays an important role in informing research, diagnostic, prevention, and treatment activities for ...
Biomolecular archaeologist looks at why most of world's population has trouble digesting beverage that helped shape civilization. Milk is something of a head-scratcher for Christina Warinner. It is rich in protein, vitamins, and minerals and has served as an important food source since prehistoric times. For much of human history, milk has ...
CORALVILLE, Iowa & HEIDELBERG, Germany--(BUSINESS WIRE)-- Genomics solutions provider Integrated DNA Technologies, Inc. (IDT) and Bio-IT company Molecular Health have entered into a global multi-year agreement, starting in the U.S., to integrate their next generation sequencing (NGS) capabilities.The partnership pairs IDT's Archer NGS research assay platform with Molecular Health's variant ...
Summary. The analysis of DNA from biological material recovered in the course of a criminal investigation can provide very powerful evidence, however when there is no match between the recovered profile and a DNA database or suspect the evidence may be of limited value. The rapidly expanding field of forensic genetics research has introduced ...
(6) Ability to work and operate effectively in a fast-paced environment. 4. Legal (1) 1L of top law schools with good academic performances (2) Completion of fundamental law courses or equivalent work experience is a plus (3) Intermediate or above level of communication skills in both English and Mandarin