Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals

Huntington's disease articles from across Nature Portfolio
Huntington's disease is a hereditary neurodegenerative disorder caused by an autosomal dominant mutation. The hallmark symptom of Huntington's disease is the presence of progressive chorea (abnormal involuntary movements), which is accompanied by psychiatric symptoms and cognitive decline.
Latest Research and Reviews

Splice modulators target PMS1 to reduce somatic expansion of the Huntington’s disease-associated CAG repeat
Somatic expansion of a CAG repeat in HTT drives onset of Huntington’s disease. Using a human cell line model and splice modulators, here the authors show that PMS1 is an enhancer of CAG repeat expansion, making it a target for therapeutic intervention.
- Zachariah L. McLean
- James F. Gusella

Neuropathogenesis-on-chips for neurodegenerative diseases
This review focuses on recent advances in on-chip platforms for patient-like in vitro modeling of the pathology of neurodegenerative diseases, including Alzheimer’s, Parkinson’s, and Huntington’s diseases as well as Amyotrophic lateral sclerosis. The authors advocate for broader usage of these human-relevant models in the academic and pharmaceutical fields.
- Sarnai Amartumur
- Huong Nguyen
- Chaejeong Heo

Iterative pseudo balancing for stem cell microscopy image classification
- Adam Witmer

Attenuated huntingtin gene CAG nucleotide repeat size in individuals with Lynch syndrome
- Karin Dalene Skarping
- Larissa Arning
- Samuel Gebre-Medhin

Retinal dysfunction in Huntington’s disease mouse models concurs with local gliosis and microglia activation
- Fátima Cano-Cano
- Francisco Martín-Loro
- Luis M. Valor

Dose-dependent reduction of somatic expansions but not Htt aggregates by di-valent siRNA-mediated silencing of MSH3 in HdhQ111 mice
- Rachelle Driscoll
- Lucas Hampton
- Andreas Weihofen
News and Comment
Mitochondrial dna instability in huntington disease.

Innate immune mechanisms mediate loss of corticostriatal synapses in Huntington’s disease
By analyzing human samples and multiple mouse models of Huntington’s disease, we found that complement proteins and microglia mediate early and selective loss of corticostriatal synapses. Strategies that block this process can reduce synaptic loss, increase excitatory input to the striatum and prevent the development of cognitive deficits in mice.
Insights into the toxic effects of mutant huntingtin
A new drug to treat chorea in huntington disease.
- Heather Wood

Correcting early circuit errors
Preventing transient defects in postnatal cortical circuit function limits disease progression in a mouse model of Huntington disease.
- Katherine Whalley
FAN1 nuclease helps to delay Huntington disease
Quick links.
- Explore articles by subject
- Guide to authors
- Editorial policies

Huntington’s Study Recognized for Potential to ‘Shape Medicine’
The journal Nature Medicine has identified a phase 3 study of pridopidine as a treatment for Huntington’s disease as one of 11 clinical trials that will shape medicine in 2022 . The URMC Clinical Trials Coordination Center (CTCC) is providing global operational support for the study, which is being conducted at more than 50 sites across the U.S., Canada, the U.K., and Europe.
The journal notes that the PROOF-HD clinical trial is one of several ongoing studies of pridopidine as a potential therapy for Huntington’s, ALS, and other neurodegenerative diseases. Pridopidine is an oral small-molecule that binds and activates the Sigma-1 receptor (S1R), which is present at high levels within the brain. By activating S1R, the drug helps boost production of brain-derived neurotrophic factor, a protein with neuroprotective properties. These protein levels are found at reduced levels in people with Huntington’s disease.
The PROOF-HD study is being conducted by Prilenia, the drug’s manufacturer, and the Huntington Study Group – a global network of more than 400 investigators, coordinators, scientists, and Huntington’s disease experts. The CTCC has collaborated with the HSG on a number of clinical trials, including the First-HD study which led to the FDA’s approval of deuterated tetrabenezine for Huntington’s in 2017.
The CTCC is providing scientific, technical, logistical, and operational logistical support for the PROOF-HD study, which announced in November 2021 that it had met its enrollment goal of 480 participants and is anticipated to run through April 2023. Elise Kayson, M.S., R.N.C., A.N.P. , director of CTCC Clinical and Strategic Initiatives, is serving as project lead for the PROOF-HD study. Kayson is also co-chair of HSG.
The CTCC is part of the Center for Health + Technology and is a unique academic-based research organization with decades of experience working with industry, foundations, and governmental researchers in bringing new therapies to market for neurological disorders. Since its inception in 1987, the CTCC has played a central role in bringing seven new drugs to market to treat Parkinson’s disease, Huntington’s disease, and periodic paralysis.
- Del Monte Institute for Neuroscience
- clinical trials

- Alzheimer's disease & dementia
- Arthritis & Rheumatism
- Attention deficit disorders
- Autism spectrum disorders
- Biomedical technology
- Diseases, Conditions, Syndromes
- Endocrinology & Metabolism
- Gastroenterology
- Gerontology & Geriatrics
- Health informatics
- Inflammatory disorders
- Medical economics
- Medical research
- Medications
- Neuroscience
- Obstetrics & gynaecology
- Oncology & Cancer
- Ophthalmology
- Overweight & Obesity
- Parkinson's & Movement disorders
- Psychology & Psychiatry
- Radiology & Imaging
- Sleep disorders
- Sports medicine & Kinesiology
- Vaccination
- Breast cancer
- Cardiovascular disease
- Chronic obstructive pulmonary disease
- Colon cancer
- Coronary artery disease
- Heart attack
- Heart disease
- High blood pressure
- Kidney disease
- Lung cancer
- Multiple sclerosis
- Myocardial infarction
- Ovarian cancer
- Post traumatic stress disorder
- Rheumatoid arthritis
- Schizophrenia
- Skin cancer
- Type 2 diabetes
- Full List »
share this!
June 7, 2023
This article has been reviewed according to Science X's editorial process and policies . Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
Promising new research on Huntington's disease zeros in on transporter protein
by University of Ottawa

New research led by a University of Ottawa Faculty of Medicine team is providing compelling insights into the mechanisms underlying the progression of Huntington's disease in an animal model. The results could lead to a greater understanding of the harrowing neurological disease in humans and help pave the way for viable drug targets and treatment approaches.
That's potentially very significant because there are presently no drugs to slow or stop the progression of the genetic brain disorder that occurs at a rate of about 1 in every 10,000 people. Huntington's disease (HD) gradually breaks down neurons in areas of the brain, progressively ravaging a patient's mind and spurring involuntary movements until sufferers are unable to walk, communicate, or even swallow. It can be passed from parent to child, typically becoming evident in middle age.
The study, published in The Journal of Neuroscience , focuses on a transporter protein referred to as VGLUT3. In the brain, this tiny protein packages glutamate into vesicles for release from neurons. Glutamate is an excitatory neurotransmitter that is involved in the most complex brain circuits. There needs to be a balance of glutamate for your brain to function properly; too much of it is associated with Huntington's and other neurological ailments.
Over the span of years, researchers led by Dr. Stephen Ferguson discovered that VGLUT3 plays a surprisingly vital role in modulating the development of Huntington's disease in the gold-standard mouse model. They bred so-called "knockout" mice that lack the transporter protein with mutant "huntingtin" mice so they could run comparisons to unveil animal models of the rare disease in both male and female mice.
Individuals diagnosed with Huntington's disease accumulate a specific mutated form of the "huntingtin" protein. This scaffolding protein is found in cells throughout the body, but the genetic defect that produces a mutant version appears to only impact the brain. The mutant triggers cell death.
Results showing the disease-modifying capacity of the VGLUT3 transporter protein were "quite remarkable," says Dr. Ferguson, a prominent professor at the uOttawa Faculty of Medicine's Department of Cellular and Molecular Medicine and Distinguished Research Chair in Neurodegeneration.
"We saw a complete reversal of Huntington disease progression in mutant huntingtin mice lacking VGLUT3," he says. "From 6 to 15 months of age, the knockout mice behaviorally were indistinguishable from wild-type mice, whereas the Huntington's mice continued to be more and more impaired over time on the various motor behavior and cognitive tasks that we tested on."
The only aspect of the symptom progression that didn't show reversal in the mouse model was anxiety behavior. But this too could prove significant because the transporter protein—which has been shown to regulate conditions such as eating disorders and drug addiction—is likely also involved in anxiety and depression.
One of the paper's reviewers described the overall results as a "substantive contribution" that "should be of wide interest to researchers in HD as well as those studying the role of VGLUT3 in cognition and motor control."
The uOttawa-led study was also chosen to be highlighted in a special feature section of the Journal of Neuroscience .
The publication's first author is Dr. Karim Ibrahim, a member of Dr. Ferguson's lab who is a newly minted Ph.D. at uOttawa. In recent years, he methodically conducted a range of behavioral experiments to generate the study's data. This included rotarod tests—one of the classic tests of motor skills in mice—and a horizontal ladder test that clearly exposed some of the impairments in the Huntington's mouse model as the animals tried to traverse it.
Efforts to develop drug targets and treatment approaches for HD must take into consideration that the "huntingtin" protein is widely expressed in the body.
"You don't really want to knock down the wild-type copy of the huntingtin gene if you can avoid it because the huntingtin protein is absolutely essential. You're better off finding a way of tricking the brain into using its circuitry slightly differently so that you can reestablish motor coordination," Dr. Ferguson says.
Ultimately, that's the goal for his lab and its collaborators in their Huntington's disease efforts. They are working on a toolkit for the pharmacological suppression of the VGLUT3 protein and exploring ways of potentially altering glutamate release in specific subsets of neurons.
"We've shown that if you block glutamate release through the activation of presynaptic receptors, that you can get an amelioration of Huntington's disease. So it may be that it will eventually require two or three different drugs to effectively treat the disease ," he says.
Explore further
Feedback to editors

Occupations that are cognitively stimulating may be protective against later-life dementia
Apr 20, 2024

Researchers develop a new way to safely boost immune cells to fight cancer
Apr 19, 2024

New compound from blessed thistle may promote functional nerve regeneration

New research defines specific genomic changes associated with the transmissibility of the mpox virus

New study confirms community pharmacies can help people quit smoking

Researchers discover glial hyper-drive for triggering epileptic seizures

Deeper dive into the gut microbiome shows changes linked to body weight

A new therapeutic target for traumatic brain injury

Dozens of COVID virus mutations arose in man with longest known case, research finds

Researchers explore causal machine learning, a new advancement for AI in health care
Related stories.

Uncovering the causes of neuron dysfunction in Huntington's disease
Apr 17, 2023

Researchers find new altered neural circuits in Huntington's disease
Jun 7, 2023

New roles found for Huntington's disease protein
Jan 21, 2020

Potential therapeutic target for Huntington's disease
Aug 16, 2016

Researchers find a protein involved in Huntington's disease motor deficits
Sep 24, 2020

Establishing a novel strategy to tackle Huntington's disease
Sep 2, 2022
Recommended for you

How myeloid cell replacement could help treat autoimmune encephalomyelitis

Researchers discover dynamic DNA structures that regulate the formation of memory

Analyzing the progression in retinal thickness could predict cognitive progression in Parkinson's patients
Let us know if there is a problem with our content.
Use this form if you have come across a typo, inaccuracy or would like to send an edit request for the content on this page. For general inquiries, please use our contact form . For general feedback, use the public comments section below (please adhere to guidelines ).
Please select the most appropriate category to facilitate processing of your request
Thank you for taking time to provide your feedback to the editors.
Your feedback is important to us. However, we do not guarantee individual replies due to the high volume of messages.
E-mail the story
Your email address is used only to let the recipient know who sent the email. Neither your address nor the recipient's address will be used for any other purpose. The information you enter will appear in your e-mail message and is not retained by Medical Xpress in any form.
Newsletter sign up
Get weekly and/or daily updates delivered to your inbox. You can unsubscribe at any time and we'll never share your details to third parties.
More information Privacy policy
Donate and enjoy an ad-free experience
We keep our content available to everyone. Consider supporting Science X's mission by getting a premium account.
E-mail newsletter
- Research at Stowers
- Technology Centers
- Publications
- Education & Outreach Education & Outreach
- Postdoc Community at Stowers
- Our Research Campus
- Our Satellite Lab at MBL
- Kansas City Community
- History & Mission
- American Century Investments
- Stowers Resource Management
- Leadership & Member Support
- For Journalists
Press Release
New research reveals the start of Huntington’s disease
Stowers scientists deduce the initiating structure of the amyloid implicated in Huntington’s and present a potential therapeutic treatment approach.
13 June 2023
Randal Halfmann, Ph.D., discusses new research revealing the start of Huntington's disease
KANSAS CITY, MO—June 13, 2023—Devastating neurodegenerative diseases like Huntington’s, Alzheimer’s, and Parkinson’s are all associated with protein deposits in the brain, known as amyloid. Despite extensive research investment into the cause and toxicity of amyloids, deciphering the first step in formation along with effective therapies has remained elusive.
For the first time, scientists at the Stowers Institute for Medical Research have uncovered the structure of the first step in amyloid formation, called the nucleus, for Huntington’s disease. The study published in eLife on June 13, 2023, from the lab of Associate Investigator Randal Halfmann, Ph.D. , proposes a new, radical method for treating not only Huntington’s but potentially dozens of other amyloid-associated diseases—preventing the initial, rate-limiting step from occurring.
“This is the first time anyone has experimentally determined the structure of an amyloid nucleus even though most major neurodegenerative diseases are associated with amyloids,” said Halfmann. “One of the big mysteries of Huntington’s, Alzheimer’s, and ALS is why disease coincides with amyloid, yet the amyloids themselves are not the main culprits.”
Fluorescence lifetime micrograph of a fluorescently tagged human protein inside yeast cells. Different colors indicate different states of protein aggregation.
Co-first authors Tej Kandola, Ph.D., and Shriram Venkatesan, Ph.D., uniquely identified the structure of the amyloid nucleus for huntingtin, the protein responsible for Huntington’s disease, discovering that the nucleus forms within a single protein molecule.
Proteins are the cell’s factory workers constructed from unique sequences of 20 amino acids, their building blocks. Some proteins have repeats of one of these amino acids—glutamine (abbreviated as Q). Huntington’s and eight other diseases, collectively called “PolyQ diseases,” occur when certain proteins have a repeat that is too long. Somehow, this causes the proteins to fold into a specific structure that starts a chain reaction that kills the cell.
“For three decades, we’ve known that Huntington’s and related fatal diseases occur when proteins contain more than around 36 Qs in a row, causing them to form chains of proteins in the brain, but we didn’t know why,” said Halfmann. “We’ve now figured out what the first link in the chain looks like, and, in doing so, have discovered a new way to stop it.”
“I am, frankly, astonished that such an intuitive physical model of nucleation emerged despite the intrinsic complexity of the cellular environment,” said Professor Jeremy Schmit, Ph.D. , from Kansas State University. “I am truly excited by the intuition and the testable hypotheses that this work inspires.”
Graphical illustration of the polyQ nucleus. Glutamines (Q) interdigitate between two antiparallel two-stranded beta sheets.
A paradigm shift and potential therapeutic method
These new findings are potentially a paradigm shift for how we view amyloids. The results from this research suggest that it is the early committed steps of amyloid formation, right after the nucleus forms, that cause neuronal cell death.
Along with uncovering the key structure that begins polyQ amyloid formation, researchers found that it only formed in isolated molecules of the protein. Clumping the proteins together in cells stopped amyloids forming altogether. This is a novel therapeutic avenue the team plans to explore further in mice and brain organoids.
A new technique
A technique recently developed by the Halfmann Lab, Distributed Amphifluoric Förster Resonance Energy Transfer (DAmFRET), shows how a protein self-assembles in single cells. This method turned out to be crucial for observing the rate-limiting amyloid-forming nucleation event.
“A key innovation was to minimize the volume of the reaction to such an extent that we can witness its stochasticity, or randomness, and then we tweak the sequence to figure out what is governing that,” said Halfmann.
Designing and testing specific patterns of Qs enabled the team to deduce the minimum structure that could form amyloid—a bundle of four strands each with three Qs in specific locations. This tiny crystal inside a single molecule of the protein is the first step in a chain reaction that results in disease.
DAmFret plots illustrating the extent of glutamine (Q) self-aggregation as the number of sequential Qs increases (left: Qs less than 36; center: Qs around 50; right: Qs over 100).
“Prior work in test tubes supports a monomeric nucleus, but this model has been controversial,” said Halfmann. “We now have strong evidence that 36 Qs is the critical number for nucleation to happen in single protein molecules, and moreover, that this is how it happens inside living cells.”
In essence, this work provides a molecular model to investigate the structure of any amyloid nucleus. Additionally, the correlation between aging and amyloids suggests that this method may ultimately uncover molecular mechanisms that cause aging. The preemptive approach to eliminate or at the very least to delay nucleation provides hope for people with pathologic PolyQ proteins.
“The emerging paradigm is that everything follows from a single event, a spontaneous change in protein shape,” said Halfmann. “That event ignites the chain reaction for amyloids that kill cells and may provide critical insight into how amyloids cause disease.”
Additional authors include Jiahui Zhang, Ph.D., Brooklyn Lerbakken, Alex Von Schulze, Ph.D., Jillian F Blanck, Jianzheng Wu, Ph.D., Jay Unruh, Ph.D., Paula Berry, Jeffery Lange, Ph.D., Andrew Box, Malcolm Cook, and Celeste Sagui, Ph.D.
This work was funded by the National Institute of General Medical Sciences of the National Institutes of Health (NIH) (award: R01GM130927) and by institutional support from the Stowers Institute for Medical Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
About the Stowers Institute for Medical Research
Founded in 1994 through the generosity of Jim Stowers, founder of American Century Investments, and his wife, Virginia, the Stowers Institute for Medical Research is a non-profit, biomedical research organization with a focus on foundational research. Its mission is to expand our understanding of the secrets of life and improve life’s quality through innovative approaches to the causes, treatment, and prevention of diseases.
The Institute consists of 20 independent research programs. Of the approximately 500 members, over 370 are scientific staff that include principal investigators, technology center directors, postdoctoral scientists, graduate students, and technical support staff. Learn more about the Institute at www.stowers.org and about its graduate program at www.stowers.org/gradschool .
Media Contact: Joe Chiodo, Head of Media Relations 724.462.8529 [email protected]
Related News
22 June 2022
The switch-like nature of the immune system
Immune cells respond to danger in an “all-or-none” fashion via protein aggregation
Read Article
In The News
03 January 2022
Stowers Investigator Randal Halfmann provides insight on prion research
The Scientist
17 March 2023
#StowersImpact: Overland Park woman honors mother, raises awareness for foundational research in the fight against Alzheimer’s
As part of the #StowersImpact campaign, the Stowers Institute for Medical Research teamed up with the Alzheimer’s Association Heart of America and Greater Missouri Chapter to put a face to the disease that impacts so many people.
Investigating the evolution of animal developmental mechanisms shows how some of Earth's earliest animals evolved
How did ancient animals develop from egg to adult, and to what extent have the genetic mechanisms that guide embryonic development endured across millennia?
Newsletter & Alerts
Read article
- April 11, 2024 | Double Trouble: Decoding the Pain-Depression Feedback Loop
- April 11, 2024 | Elemental Surprise: Physicists Discover a New Quantum State
- April 11, 2024 | A Real Life Eye of Sauron? New Technology To Detect Airborne Threats Instantly
- April 11, 2024 | This Math Problem Stumped Scientists for Almost a Century – Two Mathematicians Have Finally Solved It
- April 11, 2024 | Galactic Genesis Unveiled: JWST Witnesses the Dawn of Starlight
Huntington’s Disease Breakthrough: New Altered Neural Circuits Discovered
By University of Barcelona June 21, 2023

A recent study has discovered new alterations in neural circuits, specifically the M2 cortex’s different axonal projections to the superior colliculus (SC), in mouse models of Huntington’s disease. This finding, alongside observed reduced functional connectivity in the brain, could provide crucial data for understanding the symptoms of Huntington’s disease and for developing therapeutic approaches.
A hereditary condition that affects the brain’s neurons.
Huntington’s disease is a genetic neurodegenerative condition that results in motor, cognitive, and psychiatric impairments in those afflicted. Grasping the changes in the brain’s neural pathways in this disorder is crucial for developing therapeutic strategies. The disease has been linked to the malfunctioning of certain neuronal pathways, particularly the corticostriatal circuitry, in patients.
Now, a study published in the Journal of Neuroscience has discovered further alterations in other neural circuits using mouse models to study this pathology, which profoundly impacts the patients’ lives.
The study was led by Mercè Masana, lecturer at the Faculty of Medicine and Health Sciences of the University of Barcelona and member of the UB Institute of Neurosciences (UBneuro), the August Pi i Sunyer Biomedical Research Institute (IDIBAPS), and the Biomedical Research Networking Center on Neurodegenerative Diseases (CIBERNED). The study, whose first author is the researcher Sara Conde Berriozabal, includes the participation of the experts Jordi Alberch, Manuel José Rodríguez, and Guadalupe Soria (UB, UBneuro, IDIBAPS), among others. The study has been carried out with the support from the UB Scientific and Technological Centers (CCiTUB) and the IDIBAPS Magnetic Resonance Imaging Unit.

The administration of fluorescent sensors in the M2 cortex (in yellow) has made it possible to understand how the aberrant activity in this cortex is related to alterations in the integration of visual stimuli. Credit: University of Barcelona
An inherited disorder that affects neurons in the brain
Huntington’s disease is a rare, inherited disease that usually manifests in adults aged between 35 and 50, although there are also some juvenile forms of the disease. It is caused by a mutation in the gene called IT15 or HTT , which codes for huntingtin protein (HTT). Historically, the motor disorder that was most commonly associated with the disorder was chorea —which causes abnormal, involuntary movements— but there are also other non-motor disorders that often appear earlier.
This disorder is associated with dysfunction of corticobasal circuits in the brain. In a previous study, published in the journal eLife (2020), the team characterized one of the neural circuits involved in the development of the disease in animal models: the connection from the secondary motor cortex (M2) to the dorsolateral striatum nucleus (DSL).
In patients, the most affected brain area from the beginning of the disease is the premotor cortex —the M2 cortex in mice— which is involved in cognitive functions and perceptual processes. In the case of animal models, the M2 is associated with motor learning deficits. Moreover, this cortical area is known to be able to project neuronal axons to various brain regions beyond the striatum nucleus.

The authors of the work are members of the Neuronal Network Dysfunction Research Group in Neurological and Psychiatric Disorders of the Institute of Neurosciences of the University of Barcelona (UBneuro). Credit: University of Barcelona
Now, this study has identified for the first time that the M2 cortex sends different axonal projections to another anatomical structure in the brain —the superior colliculus (SC). These projections are deeply impaired and could be linked to the disease symptomatology.
As part of the study, the functional magnetic resonance imaging revealed the reduced functional connectivity between the left M2 cortex and all the brain regions analyzed in mice models of the disease. By applying other innovative methodologies to monitor and modulate neural activity —ontogeny, electrophysiology, photometry, and chemogenetics— the team discovered that the lack of M2 cortex activity could be responsible for the altered responses in Huntington’s disease.
Understanding the alterations in brain circuitry
Identifying the different alterations and functions of the M2 cortex circuitry —beyond the cortico-striatal pathway— provides data that are crucial to further analyze the symptoms of Huntington’s disease and other neurodegenerative pathologies (Parkinson’s disease, etc.). Also, a deeper understanding of the role of the superior colliculus and its neural circuits —involved in many neurological disorders such as Huntington’s— may provide new insights into delaying the onset and severity of the symptoms in motor disorders.
Reference: “M2 Cortex Circuitry and Sensory-Induced Behavioral Alterations in Huntington’s Disease: Role of Superior Colliculus” by Sara Conde-Berriozabal, Lia García-Gilabert, Esther García-García, Laia Sitjà-Roqueta, Xavier López-Gil, Emma Muñoz-Moreno, Mehdi Boutagouga Boudjadja, Guadalupe Soria, Manuel J Rodríguez, Jordi Alberch and Mercè Masana, 3 May 2023, Journal of Neuroscience . DOI: 10.1523/JNEUROSCI.1172-22.2023
More on SciTechDaily

Neuroscientists Built an Ultra Detailed Map of the Brain Motor Cortex, From Mice to Monkeys to Humans

Reversing Depression and Motor Dysfunction: Three Distinct Brain Circuits Contribute to Parkinson’s Symptoms

New Computer Neural Networks Identify As Well As The Primate Brain

New Map Highlights the Brain Circuits Associated With Mania

Synthetic Antioxidant Suppresses Symptoms of Huntington’s Disease in Mice

Locomotion Restored in Mice With Huntington’s-Like Condition

Human Brain Signals Recorded in Record-Breaking Resolution by New Sensor Grids

Neural Vulnerability in Huntington’s Disease Tied to Immune Response to Genetic Material
Be the first to comment on "huntington’s disease breakthrough: new altered neural circuits discovered", leave a comment cancel reply.
Email address is optional. If provided, your email will not be published or shared.
Save my name, email, and website in this browser for the next time I comment.
This Week In Huntington's Disease Research keeps you up-to-date on HDSA research activities, recently published work about Huntington’s disease, historical moments in HD research and more.
SAGE-718 on Help4HD, TEVA on AUSTEDO, PolyQ research
Posted on april 18, 2024.
Help4HD discusses SAGE-718 Cognitive changes are a frequent early symptom of HD that can have major impacts on quality of…
Tracks of HD tears, Genetic testing impacts, HD history
Posted on april 11, 2024.
Tracks of HD Tears: HD Buzz on new markers of HD progression Researchers from Germany and the Netherlands have been…
Talk on Tau, Research Webinar, HD on TEDxBath Stage
Posted on april 4, 2024.
The Talk on Tau: Cognitive changes in HD The onset of HD symptoms varies; even individuals with the same number…
2024 Human Biology Project Launch, HD History, The Buzz on CAG’s
Posted on march 28, 2024.
HDSA Launches 2024 HD Human Biology Project Letter of Intent HDSA is proud to announce the 2024 Request for Proposals…
This Week in HD Research, Survey Opportunities
Posted on march 21, 2024.
This Week in HD Research: Discovery of the HD Gene and HD Gratitude Day On March 26th, 1993, the discovery…
Pridopidine update, HDBuzz on Brains, Interview Study
Posted on march 14, 2024.
Update from Prilenia: Pursuing pridopidine for HD Earlier this week, Prilenia issued a press release and community letter announcing…
CHDI Updates: Experts and Advocates; Research Webinar
Posted on march 7, 2024.
HDBuzz Updates from the 2024 HD Therapeutics Conference Last week, from February 26th – March 1st, HD researchers from…
Rare Disease Day, CHDI, Convention Scholarships
Posted on february 22, 2024.
Celebrate Rare Disease Day with HDSA on February 29th When you have a rare disease, you face two battles –…
2024 HDSA COE Awards, CRISPR News, Research Webinar
Posted on february 15, 2024.
HDSA Names 57 Centers of Excellence and 11 Partner Sites in 2024 This week, HDSA announced its Centers of…
Convention Scholarships, Research Webinar, DNA Repair
Posted on february 8, 2024.
HDSA Convention Scholarship Applications due March 1st Join HDSA in Spokane, Washington from May 30th – June 1st for…
Research Webinar with uniQure, Online Surveys
Posted on february 1, 2024.
Upcoming Research Webinar: uniQure discusses AMT-130 Join HDSA on February 13th for a research webinar in which uniQure will present…
Neurocrine on INGREZZA, HD-CHARGE, HDBuzz on family planning
Posted on january 18, 2024.
Neurocrine discusses INGREZZA with HDSA Last month, HDSA hosted a research webinar with Neurocrine Biosciences, Inc. to discuss INGREZZA, their new…
Research Participation, Trial Stats, Upcoming Webinar
Posted on january 11, 2024.
Interested in HD Research? Get Connected with Opportunities to Participate! If you’re interested in learning more in 2024 about HD…
HDSA Convention, HDSA-funded research spotlight, New research partnerships
Posted on january 4, 2024.
Registration for HDSA’s 39th Annual Convention is now open! Join HDSA in Spokane, Washington, from May 30-June 1, 2024 for…
HDSA Research Fellowships, Clinical Trial Updates, Survey
Posted on december 21, 2023.
Applications open for the HDSA Berman-Topper and Donald King Fellowships HDSA has launched applications for the 2024 cycles of its…
Tominersen development continues, Research participation
Posted on december 14, 2023.
Continued development of Roche’s huntingtin-lowering drug tominersen Last week, Roche published the results of the GENERATION HD1 study that closed…
Research Fellowships, The Marker, Somatic Expansion
Posted on december 7, 2023.
HDSA Awards Six HD Human Biology Project Research Fellowships for 2023 After thoughtful deliberation by its Scientific Advisory Board,…
Research Participation Opportunities & Upcoming Webinar
Posted on november 30, 2023.
Online Surveys: Participate in HD Research from home! Online surveys are a great way to make your voice heard…
Giving Tuesday, HD Out-of-the-Box, Somatic Instability
Posted on november 23, 2023.
Celebrate Giving Tuesday with HDSA! At HDSA, we’re grateful for your commitment to HDSA’s vision of a world free of…
VICO Therapeutics, INGREZZA update, HD Faculty Opening
Posted on november 16, 2023.
VICO Therapeutics on Help4HD Podcast Last week on the Help4HD podcast, host Lauren Holder was joined by Dr. Katja…

Share a link
Why is it so difficult to find a treatment for Huntington’s Disease?
Health care providers may suggest certain supplements for HD patients, based perhaps on a deficiency (vitamins C, B12, E) in the blood, or for general health. But the new findings are different. The researchers didn’t set out to detect a vitamin deficiency, but instead probed the messaging within cells in the HD brain, which led them to a biochemical juncture that revealed the thiamine/biotin connection.
Genetic circuitry
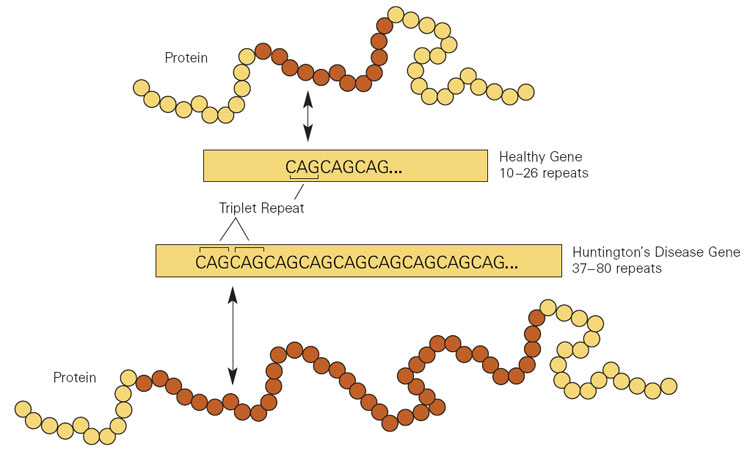
In HD, extra copies of a DNA triplet, in a gene (called Htt ) that encodes the protein huntingtin, disturb the brain’s striatum, causing the characteristic uncontrollable movements and mood manifestations. The effects arise both from the repeats in the mRNA transcribed from the expanded gene, and from what that mRNA instructs the cell to produce: extra copies of the amino acid glutamine. Long glutamine strings, called “polyQ,” are common to several brain disorders, including the spinocerebellar ataxias and spinal-bulbar muscular atrophy. (Q is the abbreviation for glutamine.)
The researchers sought genes that, when mutant or abnormally expressed, affect both the mRNA and polyQ aberrations in the Htt gene behind HD. That led to a class of genes called CPEBs, for “cytoplasmic polyA element binding protein.”
Experiments in flies had shown that CPEBs control part of the Htt mRNAs called the poly A tails. The tails are stretches of the RNA base adenine (A) at the start that stabilize the mRNA, assisting its exit from the nucleus and protecting it from being chewed up by enzymes. Nearly all mRNAs have poly A tails. A second line of evidence implicating CPEB genes is that one of them controls the amounts of other proteins that are altered in the HD brain.

Next, the new experiments.
The researchers assessed the levels of four CPEB mRNAs (aka transcripts) in striatum slices from people who had died of HD and from control brains. And they found triple the level of CPEB1 and half the normal level of CPEB4 in HD brains.
Next, findings in two mouse models echoed the human brain probings. R 6/1 mice, which have the first coding portion of the human Htt gene, exhibited the same CPEB perturbations. A second mouse model that corresponds to people who are “pre-manifest” – they have the mutation but have not yet developed symptoms – showed only the lowering of CPEB4. Perhaps the hike in CPEB1 didn’t happen because these mice don’t live long enough to show symptoms – in rodents that’s the ability to stay upright on a moving tube called a rotarod. These findings mean that CPEB4 levels plunge before CPEB1 levels triple.
When the researchers looked at the genes whose mRNAs the CPEB mRNAs control, they found three with elongated polyA tails – and the expression of this gene trio is also altered in Alzheimer’s disease and Parkinson’s disease. But it was a gene whose mRNA has a shorter polyA tail that brought vitamins unexpectedly into the picture: SLC19A3.
Mutations in SLC19A3 were already known to cause a brain disease that is treatable with biotin and thiamine! Biotin-thiamine-responsive basal ganglia disease (BTBGD) alters the protein that transports thiamine into cells. The condition causes lethargy, irritability, tremors, and spastic and uncontrollable movements, similar to HD. BTBGD arises from direct mutations in the thiamine transporter, whereas HD alters expression of the gene encoding the transporter (how rapidly it is transcribed into mRNA).
Adding high doses of biotin to the diet of people with BTBGD speeds transcription of SLC19A3, and that increases the levels of the protein that escorts thiamine into cells. But the thiamine deficiency is in the cerebrospinal fluid, not in the blood – perhaps explaining why thiamine levels are ok in the bloodwork of HD patients.
The vitamin connection inspired further experiments. Slices of striatum and cortex from the brains of deceased HD patients showed lower levels of the thiamine transporter. And thiamine was lowered in the cerebrospinal fluid but not in the blood, consistent with BTBGD.
More telling was that HD mice given high doses of biotin and thiamine in their water, starting at 3 weeks of age, no longer fell off the rotarod. And the brains of the pre-manifest mice given the vitamins were okay.
More complicated than popping supplements
On the surface, a vitamin connection to a brain disease that has evaded all attempts at treatment, for decades, is wonderful news. Taking vitamin tablets is cheap, safe, easy to do, and the vitamin is distributed throughout the central nervous system. Plus, high doses of the two vitamins are known to treat a similar disease. Even though pre-manifest individuals and people who already have signs and symptoms of HD are bound to head to the drugstore to stock up on vitamins, the researchers urge caution.
“The doses of these vitamins that led to amelioration of HD-like phenotypes in mouse models are much higher than their recommended daily intake. Besides, it is important to emphasize that, for the highest assessed doses, we observed signs of toxicity in HD mice. This led us to diminish the doses administered to the mice to a point without evident side effects and with measurable positive outcome on HD-like phenotypes,” said corresponding author José J. Lucas, Ph.D, research professor at Centro de Biología Molecular-Severo Ochoa in Madrid, in an email.
So the investigators are conducting an open label pilot clinical trial with a limited number of HD patients, to “analyze whether the high doses of thiamine and biotin required for the therapeutic effect observed in mice are safe and tolerable for patients with Huntington’s disease.” If all looks good, larger trials will follow. Meanwhile, “it is important that patients do not take thiamine and biotin supplements without advice by their neurologist,” warns Lucas.
Until vitamin supplements get an official go-ahead in slowing, treating, or delaying onset of HD, the community of families living with the disease, once again, and perhaps to a great extent than ever, has hope. And that’s priceless.
Thanks to Jane Mervar and Jonathan Monkemeyer for help with this post.
Ricki Lewis has a PhD in genetics and is a science writer and author of several human genetics books. She is an adjunct professor for the Alden March Bioethics Institute at Albany Medical College. Follow her at her website or Twitter @rickilewis
A version of this article was originally posted at PLOS and has been reposted here with permission. PLOS can be found on Twitter @PLOS
This article first appeared on the GLP Aug 18, 2023

GLP Podcasts & Podcast Videos More...

GLP Podcast: Anti-vax doctor claims COVID vaccines ‘shed’; Abandon milk and meat for the environment?
Glp podcast: rfk jr. recycles ‘gay frogs’ pesticide conspiracy; gmo v organic debate is over; scientist behind gene-edited twins back in the lab, videos more....

Video: Why does love make us feel so good? Examining its effect on our brains
Bees & pollinators more....

Are we facing an ‘Insect Apocalypse’ caused by ‘intensive, industrial’ farming and agricultural chemicals? The media say yes; Science says ‘no’

Dissecting claims about Monsanto suing farmers for accidentally planting patented seeds

Analysis: Do neonicotinoid and glyphosate pesticides threaten bees? A reassessment
Infographics more....

Are pesticide residues on food something to worry about?
Gmo faqs more....

Why is there controversy over GMO foods but not GMO drugs?

How are GMOs labeled around the world?

How does genetic engineering differ from conventional breeding?

Alex Jones: Right-wing conspiracy theorist stokes fear of GMOs, pesticides to sell ‘health supplements’

IARC (International Agency for Research on Cancer): Glyphosate cancer determination challenged by world consensus
Most popular.

Newsletter Subscription
- Weekly Newsletter (Wed)
- Daily Digest (Mon, Tue, Thu, Fri)
- Weekly Top Six (Sun)
- Featured Articles Only
- Human Articles Only
- Agriculture Articles Only
- All Types of Content
Get news on human & agricultural genetics and biotechnology delivered to your inbox.

Huntington's Disease
What is huntington's disease.
Huntington's disease (HD) is an inherited disorder that causes nerve cells (neurons) in parts of the brain to gradually break down and die. The disease attacks areas of the brain that help to control voluntary (intentional) movement, as well as other areas. People living with HD develop uncontrollable dance-like movements (chorea) and abnormal body postures, as well as problems with behavior, emotion, thinking, and personality.
For example, u ncontrolled movements in the person's fingers, feet, face, or torso. These movements are signs of chorea. They can get more intense when the person is nervous or distracted; as HD progresses, the person's movements can become more extreme and obvious.
Symptoms of HD typically appear in middle-aged people (adult HD). They can also appear in children (juvenile HD), but this is rare. The disease gets worse over time.
Early signs of HD can vary, but often include mild clumsiness or problems with balance or movement, cognitive or psychiatric symptoms (problems with thinking or emotion), and changes in behavior. For some people, chorea can make it harder to walk, which increases the chances of falling. Some people with HD do not develop chorea; instead, they may become rigid (stiff) and move very little or not at all. This condition is called akinesia. Other people may start out with chorea but become rigid as the disease progresses. In addition to chorea, some individuals have unusual fixed (unchanging) postures, which is known as dystonia. The two movement disorders (akinesia and dystonia) can blend or alternate. Other symptoms may include tremor (unintentional back-and-forth movement in the person's muscles) and unusual eye movements. The eye movements can happen early in the disease.
Physical changes may include slurred speech and problems with swallowing, eating, speaking, and especially walking. People with HD may lose weight because of problems with feeding, swallowing, choking, and chest infections. Other symptoms may include insomnia (having trouble sleeping), loss of energy, fatigue, and seizures. Eventually the person will need to stay in bed or in a wheelchair.
Changes in thinking (cognitive changes) may include problems with attention or judgment and having difficulty solving problems or making decisions. Other changes may include trouble with driving, prioritizing (deciding which things are more important to do and which are less important), and organizing, learning new things, remembering a fact, putting thoughts into words, or answering a question. These cognitive changes get worse as the disease progresses, until people with HD are not able to work, drive, or care for themselves. When the cognitive problems are severe enough that the person cannot function in daily life, the condition is described as dementia. But many people with HD stay aware of their environment and can express their emotions.
Changes in behavior may include mood swings; feeling irritable (cranky); not being active; or feeling apathetic (uninterested), depressed, or angry. These symptoms may decrease as the disease progresses. But in some people, the symptoms can continue and may include angry outbursts, thoughts of suicide, deep depression, and psychosis (losing touch with reality). People with HD may withdrawal from social activities.
Who is more likely to get Huntington's disease?
HD is an inherited disorder. It is passed from parent to child through a mutation (a change) in a particular gene. When a parent has HD, each child has a 50% chance of inheriting the copy of chromosome 4 that carries the HD mutation. If a child does not inherit the HD mutation, he or she will not develop the disease and cannot pass it on to future generations. When HD occurs without a family history, it is called sporadic HD.
HD is caused by a mutation in the gene for a protein called huntingtin. The defect causes the building blocks of DNA called cytosine, adenine, and guanine (CAG) to repeat many more times than they normally do.
Most people have fewer than 27 CAG repeats in their HD gene, so they are not at risk for the disease. People who have CAG repeats in the middle range (27 to 35) are not likely to develop the disease, but they could still pass it on to future generations. People with HD may have 36 or more CAG repeats.
Each child of a parent with HD has a 50% chance of inheriting the HD gene. A child who does not inherit the HD gene will not develop the disease, and generally, they cannot pass it on to their children or other future generations.
How is Huntington's disease diagnosed and treated?
Diagnosing HD
In general, doctors use a combination of tests and other information to see if a person has HD. These include medical history, neurological and lab tests, brain imaging, and genetic testing.
- Neurological exam and medical history—A neurologist will conduct an in-depth interview to obtain a medical and family history for the individual and to rule out other conditions. Neurological and physical exams may review reflexes, balance, movement, muscle tone, hearing, walking, and mental status. Laboratory tests may also be ordered, and individuals with HD may be referred to specialists such as psychiatrists, genetic counselors, clinical neuropsychologists, or speech pathologists for specialized management and/or to support diagnosis.
- Diagnostic imaging—In some cases, especially if a person's family history and genetic testing are inconclusive, the physician may recommend brain imaging, such as computed tomography (CT) or, more likely, magnetic resonance imaging (MRI). As the disease progresses, these scans typically reveal shrinkage in parts of the brain and enlargement of fluid-filled cavities within the brain called ventricles. These changes do not necessarily indicate HD, because they can occur in other disorders. A person can have early symptoms of HD and still have normal findings on a CT or MRI scan.
- Genetic tests—Genetic testing can confirm or rule out a suspected genetic condition or help determine a person's chance of developing or passing on a genetic disorder. Genetic testing makes it possible to predict with a higher degree of certainty if someone will develop HD. The most effective and accurate method of testing for HD—called the direct genetic test—counts the number of CAG repeats in the HD gene, using DNA taken from a blood sample. The presence of 36 or more repeats supports a diagnosis of HD. A test result of 26 or fewer repeats rules out HD. Prenatal testing is an option for people who have a family history of HD and are concerned about passing the disease to a child.
Treating HD
There is no treatment that can stop or reverse HD, but some of the symptoms can be treated:
- The drugs tetrabenazine and deuterabenazine can treat chorea associated with HD
- Antipsychotic drugs may ease chorea and help to control hallucinations, delusions, and violent outbursts. Some antipsychotic medications can have side effects that make muscle contraction symptoms of HD worse. Individuals using antipsychotic drugs for Huntington's disease symptoms should be closely monitored for side effects.
- Drugs may be prescribed to treat depression and anxiety
Side effects of drugs used to treat the symptoms of HD may include fatigue, sedation, decreased concentration, restlessness, or hyperexcitability. These drugs should be only used when HD symptoms create problems for the person living with HD.
What are the latest updates on Huntington's disease?
Researchers are learning more about Huntington's disease over time. Below are some important updates that may improve how doctors care for this disorder in the future.
Understanding Huntington's disease mechanisms
NINDS-funded researchers are trying to better understand the cellular and molecular mechanisms involved in HD by investigating, for instance, how the huntintin protein affects cell signaling and how its altered structure can contribute to disease. The following provides an overview of this research:
- A new avenue of NINDS-supported research is asking whether additional changes to the Huntington gene during development and in adulthood impact disease onset and severity, and whether the Huntington gene affects the brain's overall ability to maintain healthy, undamaged DNA. This work is a promising area for identifying new modifiers of HD onset and progression that may be attractive drug targets.
- Excessive chemical signaling between cells in the brain may lead to chronic overexcitation (overactivation of neurons to turn on), which is toxic to neurons. Several labs are investigating whether drugs that counteract excitotoxicity might help against HD.
- Cutting-edge methods such as optogenetics (where neurons are activated or silenced in the brains of living animals using light beams) are being used to probe the cause and progression of cell circuit defects in HD.
The NINDS-funded PREDICT-HD study and several international studies are working to identify and validate biomarkers for HD. Biomarkers are biological indicators that can be used to predict, diagnose, or monitor a disease. One goal of PREDICT-HD is to determine if the progression of the disease correlates with changes in brain scan images, or with chemical changes in blood, urine, or cerebrospinal fluid. Another goal is to find measurable changes in personality, mood, and cognition that typically precede the appearance of motor symptoms of HD. A third phase of PredictHD is ongoing.
A related NINDS-supported study aims to identify additional human genetic factors that influence the course of the disease. Finding genetic variants that slow or accelerate the pace of disease progression promises to provide important new targets for disease intervention and therapy.
Through a NINDS-funded consortium, researchers are using cultures of cell lines (created from people with HD who have donated skin and blood samples for research) to understand why neurons malfunction and die in HD, and to rapidly test potential new drugs. Another approach may be to mobilize stem cells that are already there and can move into damaged tissue.
Turning research into treatment
Testing investigational drugs may lead to new treatments and at the same time improve our understanding of the disease process in HD. Classes of drugs being tested include those designed to control symptoms, slow the rate of progression of HD, block the effects of excitotoxins, provide support factors that improve neuronal health, or suppress metabolic defects that contribute to the development and progression of HD.
Several groups of scientists are using gene-editing or specific molecules that can interfere with the production of the Huntingtin protein in cells or animals to stop production of Htt in inappropriate locations or amounts.
Scientists are using imaging technology to learn how HD affects the chemical systems of the brain, characterize neurons that have died, view changes in the volume and structures of the brain in people with HD, and to understand how HD affects the functioning of different brain regions.
Brain development
Altered brain development may play an important role in HD. Huntingtin is expressed during embryonic development and throughout life. Studies in animals have shown that the normal HD gene is vital for brain development. Adults who carry the mutant HD gene but have not yet displayed symptoms show measurable changes in the structure of their brain, even up to 20 years before clinical diagnosis.
A NINDS-funded study is evaluating brain structure and function in children, adolescents, and young adults up to age 30 who are at risk for developing the disease because they have a parent or grandparent with HD. This study is trying to capture potential HD effects during the late stages of brain development. Participants who carry the expanded gene will be compared to individuals who carry the gene but have fewer than 9 CAG repeats, as well as to individuals who do not have a history of HD in their family. Changes in brain structure and/or function in the gene-expanded group may point to a developmental component in HD.
How can I or my loved one help improve care for people with Huntington's disease?
Consider participating in a clinical trial so clinicians and scientists can learn more about HD. Clinical research uses human volunteers to help researchers learn more about a disorder and perhaps find better ways to safely detect, treat, or prevent disease.
All types of volunteers are needed— those who are healthy or may have an illness or disease— of all different ages, sexes, races, and ethnicities to ensure that study results apply to as many people as possible, and that treatments will be safe and effective for everyone who will use them.
For information about participating in clinical research visit NIH Clinical Research Trials and You . Learn about clinical trials currently looking for people with HD at Clinicaltrials.gov .
Where can I find more information about Huntington's disease? Information may be available from the following organizations and resources: Hereditary Disease Foundation Phone: 212-928-2121 Huntington's Disease Society of America Phone: 212-242-1968 or 800-345-4372 National Library of Medicine Phone: 301-594-5983 or 888-346-3656
The browser you are using is no longer supported and for that reason you will not get the best experience when using our website.
You currently have JavaScript disabled in your web browser, please enable JavaScript to view our website as intended.
New network launched to advance research into Huntington’s disease
Dr Sarah Gunn and Professor Flaviano Giorgini
A new network has been launched by the University of Leicester to drive forward research, interventions and engagement for those living with Huntington’s disease.
Huntington's disease is a life-limiting inherited condition that damages nerve cells in the brain causing them to stop working properly. This leads to loss of movement and cognition over many years, as well as changes in emotions and personality.
Now it’s hoped the Leicestershire Huntington’s Disease Network (LHDN), which was officially launched on Saturday (20 April) at a special event at Attenborough Arts Centre, will help drive forward innovation and research taking place as part of a collaboration between patients, charity partners, NHS services as well as University leads.
Lecturer and practising clinical psychologist Dr Sarah Gunn and neurogenetics Professor Flaviano Giorgini from the University are running the network alongside senior staff from Leicestershire Huntington’s Disease NHS services.
Dr Gunn has been working with Huntington’s disease patients for over 10 years, helping to understand the psychological difficulties experienced by individuals alongside their family members. She has developed a number of therapeutic programmes for patients and delivers them through the Huntington’s Disease Association (HDA).
She said: “We believe this is the first network of its kind in the East Midlands to bring together people and families affected by Huntington’s disease, healthcare staff, and researchers. We’re hugely excited about the difference we could make as part of this collaboration and the benefits it could bring to patient care.
“One of the worst elements of living with Huntington’s is the feeling of isolation and hopelessness, whether as a person carrying the gene or as a family member. We aim to provide hope and solidarity. This network shows how passionate we are about improving the lives of people affected.
“The launch itself was an incredibly positive and inspiring event, which gave us an opportunity to gather thoughts and ideas for future research from people affected by Huntington’s so that we can try and build a brighter future for this severely under-served population.”
As well as learning about current research taking place at the University, those attending the launch heard from Professor Anne Rosser from the European Huntington’s Disease Network (EHDN) and Cath Stanley, chief executive of the HDA, alongside healthcare staff affiliated to clinical and genetic services.
Professor Giorgini has been researching Huntington's since 2003, using model organisms to study the pathogenic mechanisms underlying the disease with the hope of identifying potential therapeutic targets. He is also Science Director of the EHDN, where he helps coordinate research grants and the science strategy.
He said: “This new network will be critical in bringing together Huntington’s disease stakeholders, which we hope will lead to expansion of research and improvements in the quality of lives of affected individuals and their families.”
Related stories
Sleeping giant surprises gaia scientists, nhs staff retention to be investigated in new study, data with dignity: university of leicester trials new ethical surveying software, ethnic minorities are underrepresented in studies into multiple long-term health conditions, research reveals, gp numbers associated with life expectancy in england, study shows, new analysis reveals the brutal history of the winchcombe meteorite’s journey through space.
How many is too many? Exploring the toxic CAG threshold in the Huntington’s disease brain
Dr sarah hernandez | april 21, 2024, latest news.

Cry your eyes out: detecting huntingtin in tears
Is someone cutting onions? Expanded huntingtin can now be detected in tears to help scientists track disease progression.
Dr Leora Fox | April 10, 2024

The director’s cut: how CAG repeats change the editing of genetic messages
Scientists in Massachusetts have recently advanced our understanding of how repetitive sequences in DNA can disrupt the creation and editing of genetic messenger molecules in cells, and how this could lead to the production of harmful proteins.
Dr Rachel Harding | March 26, 2024

Making babies: having a family, the HD way
Making babies: HDBuzz's feature article - updated for 2024 - on fertility technologies that can help at-risk people to have HD-free children
Betony Childs and Dr Nayana Lahiri | March 16, 2024

Understanding expansions at the single cell level
Scientists have looked at CAG expansions in brains from people with HD to see which cells are affected
Dr Rachel Harding | March 12, 2024

Huntington's Disease Therapeutics Conference 2024 - Day 3
Dr rachel harding, dr leora fox, and dr sarah hernandez | march 07, 2024.

Huntington's Disease Therapeutics Conference 2024 - Day 2
Dr rachel harding, dr sarah hernandez, and dr leora fox | march 06, 2024.

Huntington's Disease Therapeutics Conference 2024 - Day 1
Check out research updates from Day 1 of the 2024 HD Therapeutics Conference #HDTC2024
Dr Rachel Harding, Dr Sarah Hernandez, and Dr Leora Fox | March 05, 2024

CRISPR-based drugs: one giant leap for mankind
Casgevy is the first CRISPR-based drug to make its way through the approval process, all but curing Sickle Cell Disease and it’s paving the way for similar drugs targeting other diseases. Is Huntington’s disease next?
Dr Rachel Harding | February 14, 2024

Steady progress from uniQure - promising data to end the year
uniQure ushered in the end of the year by releasing some promising data from their huntingtin-lowering gene therapy trials
Dr Rachel Harding | December 20, 2023

Putting it in print: GENERATION HD1 study results published
Data from GENERATION HD1, the Phase 3 clinical trial testing the huntingtin-lowering drug tominersen, have just been published in a scientific journal. The trial ended a while back, so why is this an important milestone, and what’s next?
Dr Sarah Hernandez, Dr Rachel Harding, and Dr Leora Fox | December 07, 2023

Regulating repetition: Gaining control of CAG repeats could slow progression of Huntington’s disease
Many diseases are caused by repetitive DNA sequences. Understanding the regulation of those repetitive sequences may hold the key for unlocking therapeutics for Huntington’s disease. A team from Toronto has just advanced our understanding.
Dr Jeff Carroll | November 30, 2023
Previously featured.
Dr Leora Fox
Dr Rachel Harding
Dr Rachel Harding, Dr Leora Fox, and Dr Sarah Hernandez
Dr Rachel Harding, Dr Sarah Hernandez, and Dr Leora Fox
New to HD research? Click here to get an overview
A Freedom Tower on Chromosome Five
Subscribe in iTunes
Podcast RSS Feed
Suggest an article
Is there something you want us to write about? Tell us about it here. We consider all suggestions but can't promise to write content about any particular suggestion.
Email [email protected]

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- v.18(1); 2023 Jan
Recent approaches on Huntington's disease (Review)
Anastasia marina palaiogeorgou.
1 Laboratory of Genetics, Department of Biotechnology, School of Applied Biology and Biotechnology, Agricultural University of Athens, 11855 Athens, Greece
Eleni Papakonstantinou
Rebecca golfinopoulou, markezina sigala, thanasis mitsis, louis papageorgiou, katerina pierouli, konstantina dragoumani, demetrios a. spandidos.
2 Laboratory of Clinical Virology, School of Medicine, University of Crete, 71003 Heraklion, Greece
Flora Bacopoulou
3 University Research Institute of Maternal and Child Health and Precision Medicine, and UNESCO Chair on Adolescent Health Care, National and Kapodistrian University of Athens, ‘Aghia Sophia’ Children's Hospital, 11527 Athens, Greece
George P. Chrousos
Elias eliopoulos, dimitrios vlachakis.
4 Division of Endocrinology and Metabolism, Center of Clinical, Experimental Surgery and Translational Research, Biomedical Research Foundation of the Academy of Athens, 11527 Athens, Greece
Associated Data
Not applicable.
Huntington's disease (HD) is a neurodegenerative disorder characterized by severe motor, cognitive and psychiatric symptoms. Patients of all ages can present with a dysfunction of the nervous system, which leads to the progressive loss of movement control and disabilities in speech, swallowing, communications, etc. The molecular basis of the disease is well-known, as HD is related to a mutated gene, a trinucleotide expansion, which encodes to the huntingtin protein. This protein is linked to neurogenesis and the loss of its function leads to neurodegenerative disorders. Although the genetic cause of the disorder has been known for decades, no effective treatment is yet available to prevent onset or to eliminate the progression of symptoms. Thus, the present review focused on the development of novel methods for the timely and accurate diagnosis of HD in an aim to aid the development of therapies which may reduce the severity of the symptoms and control their progression. The majority of the therapies include gene-silencing mechanisms of the mutated huntingtin gene aiming to suppress its expression, and the use of various substances as drugs with highly promising results. In the present review, the latest approaches on the diagnosis of HD are discussed along with the need for genetic counseling and an up-to-date presentation of the applied treatments.
1. Introduction
Neurodegenerative disorders have a exhibited a marked increase in incidence worldwide, thus rendering them a primary concern for the scientific society. The genetic cause of numerous disorders has already been described ( 1-3 ) and, nowadays, research focuses on the timely diagnosis and effective therapy of the most common neurodegenerative disorders, such as Parkinson's disease, Alzheimer's disease, amyotrophic lateral sclerosis and Huntington's disease (HD). These disorders have diverse clinical manifestations; however, some of them demonstrate similarities among patients ( 4 , 5 ). Although in numerous cases, the onset of neurodegenerative disorders appears in middle to late adult life, there are patients who manifest symptoms of these disorders at a very early age ( 6 , 7 ). HD is one of the most common disorders with severe symptomatology, which affects individuals of all ages, progressively leading to severe disabilities. The genetic basis of this disorder has been established and has been known for a few decades now, and recent research has revealed promising mechanisms for eliminating HD symptoms ( 8 ). Furthermore, HD can be regarded as a model neurodegenerative disorder for the study of other cases with shared symptoms, and knowledge of other diseases may be useful for HD diagnosis and treatment.
2. Genetics and pathology of Huntington's disease
HD is a fatal, autosomal dominant, progressive neurodegenerative disorder characterized by severe symptoms, including motor, cognitive and psychiatric symptoms, atrophy of the basal ganglia and the cerebral cortex, and an inevitably progressive course, resulting in mortality 5-20 years following the manifestation of symptoms. Typically, the motor defects include chorea and loss of coordination, and patients also demonstrate difficulty with speech and swallowing ( 9 ). Cognitive symptoms can be detected up to a decade prior to diagnosis and cognitive ability declines as the disease progresses ( 10 ). Psychiatric symptoms, such as depression, psychosis and obsessive-compulsive disorder, are also common in HD and are particularly distressing for patients ( 11 , 12 ). Patients with HD eventually require a wheelchair and more severe symptoms may lead to them becoming bedridden, with all the complications that may derive from that form of immobility.
From a neuropathological point of view, in patients with HD, the dysfunction and death of specific neurons within their brains are observed. There is a wide range in the age of onset of HD, as both juveniles ( 13 , 14 ) and adults have been diagnosed with the disorder thus far. For instance, kindred members of families that revealed a history consistent with HD autosomal dominant inheritance, took part in a 20-year study, which was published in 2004( 15 ). The researchers of that study found that the typical ages of disease onset were between 21 and 50 years of age ( 15 ). Although the disorder typically manifests in adulthood, juvenile HD (JHD) is also frequent among patients ( 16 ). A recent study, conducted in Argentina in 2015( 17 ), reported that almost 20% of the patients diagnosed with HD, revealed their first symptoms of the disorder during their childhood. It should be noted that the overall estimated prevalence of JHD of that study was higher than that in any other population recorded to date ( 17 ). The brain structure in young patients was previously assessed by Tereshchenko et al ( 18 ) in 2019, proposing that the morphology differs among juveniles and adult patients, as young patients revealed proportional cerebellar enlargement ( 18 ). In the same year, another study suggested that the pathogenesis of HD begins with abnormal brain development in both child and adolescent patients ( 19 ).
The first attempt to discover the genetic cause for HD by Gusella et al ( 20 ) revealed that the HD gene is linked to a polymorphic DNA marker that maps to human chromosome 4, in particular 4p16.3. A decade later, a Huntington's Disease Collaborative Research Group discovered that mutations in the Huntington (HTT) gene encoding the huntingtin protein, a large protein of 3,144 amino acids, led to the neurodegenerative disorder ( 21 ). In particular, they suggested that the disorder is caused due to a cytosine-adenine-guanine (CAG) trinucleotide expansion in exon 1 that codes for polyglutamine (polyQ) in the N-terminal of the HTT gene ( 21 ). The CAG sequence is normally repeated 9 to 35 times, with an average median of between 17 and 20 repeats. However, patients with HD usually reveal a CAG expansion exceeding 35 repeats ( 22 ). Above a threshold of ~35 CAG repeats, the age of onset of HD is inversely associated with the length of the expansion. A recent study conducted by Schultz et al ( 23 ) demonstrated that the development of verbal skills appeared to plateau earlier as CAG repeat length increased. The repeats are usually between 36 and 39, depending on the age. Juveniles with HD exhibit high repeat lengths ( 24 ). In certain rare cases, patients exhibit less repeats in their genome, 27-35, demonstrating an endophenotype ( 25 ).
Of note, the huntingtin protein is expressed in all cell types of the body, both at the tissue and subcellular level, in all developmental stages. Recently, research has focused on the investigation of the HTT structure via cryo-electron microscopy contributing to a better comprehension of its morphology and form abnormalities ( 26 ). It has been described as a 350-kDa HEAT-repeat protein which interacts with hundreds of other proteins ( 27 ) and participates in numerous cellular processes. Although the cellular functions of HTT protein are not yet completely understood, it appears to play a crucial role during early embryonic development and neurogenesis. In particular, Saudou and Humbert ( 22 ) described the human huntingtin protein sequence and its post-translational modifications in detail. They suggested that it coordinates cell division, as it participates in the proper mitotic spindle positioning and it regulates ciliogenesis. They also noted that huntingtin mediates endocytosis, vesicle recycling and endosomal trafficking, as it interacts with other proteins which are related to these mechanisms. Other functions of the protein include autophagy and transcription ( 22 ). It should be underlined that the HTT gene physiological expression is essential for organism homeostasis as it plays a neuroprotective role as well, even against mutant HTT (mHtt) toxicity ( 28 , 29 ). Moreover, HTT protein plays a crucial role in mitochondrial structure and function in the embryogenesis and oxidative metabolism, and HTT mutations have been linked to mitochondrial abnormalities ( 30-32 ).
The role of both wild-type Htt and mHtt in gene silencing studies has been investigated for the development of an effective therapy. As regards mHtt molecules, they form toxic aggregates into the central nervous system, depending on the length of polyQ expansion. For instance, mHtt co-aggregates with other proteins which play a crucial role in the cell, leading to misfunctioned phenotypes. Numerous studies have focused on the effects of HTT gene knockouts and knockdowns in cellular function. For example, a study published in 2017 suggested that mutations in HTT protein are related to nucleocytoplasmic transport disruption, leading to the improper function of cells ( 33 ). During initial experiments performed on mice with HTT knockdown mutants, the mice succumbed after 8 days of gestation ( 34 ). Other studies have demonstrated that HTT deletion in the mouse central nervous system leads to a phenotype similar to that of HD ( 35 , 36 ). It is worth mentioning that a recent study suggested that HTT variants are also linked to another disorder with similar symptomatology with HD, the so-called Lopes-Maciel-Rodan syndrome ( 37 ). In addition, other studies have revealed the ability of various molecular chaperones, such as the heat shock family proteins, HSP40, HSP70, HSP90 and HSP105, to combine with misfolded mHtt and inhibit aggregate formation, leading to cell survival ( 38 , 39 ).
It is clear that the loss of HTT function contributes to HD pathology and for this reason, it is essential for survival. The reduction of mHtt levels should be accompanied by regular HTT expression.
3. Diagnosis and genetic counseling
It has already been mentioned that the age of onset of HD is inversely associated with the length of the expansion in the HTT gene. For instance, rare carriers of 36 to 39 CAG repeats have lower penetrance and a later onset of the disease than those with 40 or more CAG repeats. Additionally, Keum et al ( 40 ) found that, along with clinical onset, the age of patients with HD at the time of death was well determined by an expanded CAG-repeat length. However, they claimed that the overall duration of the disease was independent of the length of the mutation's ( 40 ). These data may be useful, not only for the molecular diagnosis of the disorder, but also for the prediction of the outset of HD symptomatology. For the molecular diagnosis of the disorder, various PCR methods have been demonstrated in order to detect CAG expansions ( 41 ). A recent study presented a novel triplet-primed PCR-based assay aiming to improve the test reliability and accuracy by detecting CAG expansions in samples with sequence variations in the HTT gene ( 42 ).
It is known that miRNAs are involved in the biological processes of development, proliferation, inflammation and apoptosis, and their expression has been linked to HD diagnosis and symptomatology. For instance, Langfelder et al ( 43 ) found that the abnormal expression of miRNAs played a critical role in HD pathogenesis. For this reason, apart from the direct quantification of mHTT itself, which is the main disease-related biomarker, other miRNAs may be useful tools as biomarkers for HD prognosis ( 44 ).
Furthermore, numerous diagnostic tests have been proposed thus far, based on criteria related to inheritance and the symptomatology of the individual; however, these methods need to be improved. Patients who experience certain cognitive and behavioral symptoms may have HD ( 45 , 46 ). A recent study proposed the Enroll-HD dataset for estimating disease onset and its diagnostic confidence level ( 47 ). The results of that study were not promising, suggesting that it is important to develop more reliable diagnostic criteria ( 47 ). Another diagnostic approach suggested that the concentration of trace elements in the blood of patients with HD differs from that of healthy individuals. Researchers found increased levels of the essential elements iron, chromium, selenium and zinc and of the non-essential element, arsenic, in the blood of patients with HD, suggesting that the blood metal profile may be used as an easy tool for the disorder's medical detection ( 48 ).
HD follows the Gregor Mendel's principles of inheritance, as it is inherited in an autosomal-dominant manner. The offspring of an individual with a pathogenic variant, heterozygote, have a 50% chance of inheriting the disease-causing allele. Genetic counseling includes predictive testing in asymptomatic adults and prenatal testing in order to reveal the mutated allele ( 49 ). The prevalence of HD is ~1 in 10,000 individuals in the USA, as well as in Europe ( 50 , 51 ). In the year 2000, Sobel and Cowan ( 52 ) conducted predictive testing on asymptomatic individuals at risk of developing HD in the context of genetic counseling. Family members were requested to describe their communication and interactions with the social environment, and provide concerns about their future care. Members in 50% of the families experienced changes in patterns of communication and 56% of the participants reported changes in current relationships. The researchers suggested that families may benefit in pretest sessions by examining their patterns of dealing with illness issues, both past and present ( 52 ).
Migliore et al ( 53 ) suggested different approaches of counseling, depending on the genetic condition of the individual. For instance, in the case of intermediate alleles (27-35 CAG repeats) the experts should explain the potential risk of mutations and other members of the family should also be tested. In the case of low penetrance alleles (36-39 CAG repeats), individuals should be informed about the risk of HD symptoms manifesting at any age. Counseling for all family members is also required when juveniles are diagnosed with JHD. When the HD mutation is detected in a prenatal genetic test, the parents should be informed for the risk of the newborn manifesting the disease and should be given the option of terminating the pregnancy ( 53 ). This approach utilizes the current knowledge of the molecular basis of HD with the inclusive genetic counseling of all relatives. Recently, MacLeod et al ( 54 ) proposed a family systems approach to genetic counseling, which uses the narrative model. With the narrative resources, the genetic counselor can contribute to generate new meanings that the person may give to their experience of the genetic condition and help the patient adapt to living with the disorder or its risks ( 54 ). Another interesting comparative study, that was performed over the past two decades, on how parents inform their children who are at risk about their genetic risk demonstrated that, although testing is performed more often, the overall attitude towards information and testing has not changed significantly ( 55 ). This ascertainment proposes that new methods for more comprehensible information and accessible genetic counseling need be developed.
4. Treatment
Research focusing on understanding the underlying molecular mechanisms leading to the HTT gene mutations is highly promising, aiming to find a cure for HD. However, current treatments for HD are still limited. The therapies applied focus on the treatment of symptoms, as neuroprotective therapies to prevent disease onset and to attenuate the progression of the disease are not yet available. For instance, it has been proven that HD is caused by toxic properties of mHTT, rather than merely the decrease of wild-type HTT; for this reason, approaches focusing on mHTT expression, such as lowering HTT mRNA and mutant huntingtin protein, appear to be promising ( 56 ). The main strategies which have been demonstrated thus far as treatments for HD are presented in Fig. 1 .

Box diagram of the main strategies which have been demonstrated thus far as treatments for HD. HD, Huntington's disease; CAG, cytosine-adenine-guanine.
A recent study proposed that targeting of CAG repeat-dependent mechanisms, through gene-silencing approaches, may affect the rate of functional, motor and cognitive impairment, but not weight loss, in manifest HD mutation carriers ( 57 ). The standard approaches to DNA targeting use some form of specific DNA-binding element combined with nucleases, epigenetic modulators, or transcription factors. Zinc-finger transcriptional repressor approaches may lower mHTT levels by targeting DNA without altering it, whereas zinc-finger nucleases can add to the repressive effect of Zinc-finger proteins that reduce the levels of gene expression by simply binding to DNA and preventing gene transcription by actually disrupting or correcting the mutant gene ( 58 ). Along with similar techniques to other direct genome editing strategies, such as CRISPR/Cas9, strategies that are targeted in lowering huntingtin and HTT genome editing have immense potential for the treatment of HD. The main advantage is the permanent correction of the disease-causing CAG expansion. An antisense mechanism targeting HTT RNA, using synthetic antisense oligonucleotides (ASOs) that bind to the specific sequence of ribonucleic acid, may reduce the mRNA translation to the HTT disease-causing protein ( 59 ). The ASOs are widely distributed throughout the central nervous system and they do not require a viral or lipid carrier, resulting to an effective and simple to develop treatment. Tabrizi et al ( 60 ) used the antisense oligonucleotide IONIS-HTTRx designed to inhibit HTT mRNA by triggering the RNase H1-mediated degradation of the target mRNA, in order to minimize the concentration of mutant huntingtin in cerebrospinal fluid. They conducted an extended research with 34 patients who were treated with increasing dose levels of 10 to 120 mg and the observations were compared with individuals who received the placebo. The results revealed that the reduction in concentrations of mutant huntingtin was dose-dependent ( 60 ). The strategy for post-transcriptional gene suppression using non-coding double-stranded RNA sequences is known as the RNA interference (RNAi) mechanism. The RNAi pathway has been used thus far to suppress specific genes of interest and the results are highly promising for numerous diseases. There are various molecules which can be used for this purpose, such as siRNAs, shRNAs and artificial miRNAs that have been used to eliminate the HD symptoms. The first trials were performed two decades ago in rodents. In 2005, Harper et al ( 61 ) used a shRNA molecule to target the HTT mutant gene and the results were satisfactory, as the reduction in mHTT synthesis, prevented inclusions, gait deficits and rotarod dysfunction. In another case, a siRNA molecule was injected into the mouse striatum, and the reduction in mHTT synthesis prolonged striatal neuron survival, reduced aggregates and prevented motor dysfunction ( 62 ). The application of siRNA approaches has been successful in multiple animal systems ( 63 ). In a recent study for example, a single-stranded siRNA (ss-siRNA) was used for RNAi, resulting in a selective decrease of CAG-expanded HTT protein in various regions throughout the mouse brain ( 64 ). Other ribonucleic acids, such as miRNAs have been used for the suppression of mHTT in genetically modified mice and the results have been promising; in one case, this strategy led to the prevention of regional cortical and striatal atrophy, and reduced weight loss ( 65 ). It is worth mentioning that the majority of the previous technics, apart from gene editing, effectively reduce, but do not completely eliminate the production of mHTT.
In addition, the therapeutic approach of overexpressing wild-type HTT has been investigated. Early trials of inserting the wild-type HTT into mammalian cells which expressed mHTT have led to reduced cell death ( 26 ).
Numerous research studies have used viral vectors, such as adeno-associated virus, which encapsulate the RNA molecules, and their genome is combined with enhancers and promoters, in order to deliver these agents by injection into the body ( 66 ).
Different compounds may be candidates for the treatment of neurodegenerative disorders, including HD. A recent study proposed the utilization of flavonoids, which may reduce cellular stress and play an anti-inflammatory and anti-apoptotic role in the cell ( 67 ). In another case, researchers proposed that marine compounds may be used for the treatment of various neurodegenerative diseases, as they also demonstrate antioxidant, anti-inflammatory and anti-apoptotic properties. Tetrabenazine (TBZ), which is an inhibitor that blocks dopamine uptake into vesicles, has been shown to exert antichorea effects in patients with HD and was the first approved drug for medication ( 68 ). Since then, studies on the optimization of drug delivery and bioavailability of TBZ in patients have been conducted based on latest nanotechnology technics ( 69 ). Several molecules have been suggested for the treatment of Parkinson's disease, such as fucoidan and xyloketal B, and fucoxanthin and cerebrosides for Alzheimer's disease, and have also been investigated for other disorders, such as HD for effective treatment ( 70 ). In 2020, Jabłońska et al ( 71 ) suggested that pridopidine, a dopamine stabilizer, may be a promising drug for HD symptoms. It is well-known that there is an association between the amount of dopamine in the central nervous system and the stage of the disease, as the causes of HD are dopaminergic conduction disorders, and experiments on animal models have demonstrated the protective effect of pridopidine on nerve cells ( 72 , 73 ). Τhe main advantage of drug treatment for HD is that the effectiveness and tolerance of each active compound is well-studied for other neurodegenerative diseases with similar symptomatology. Thus, it is easier to design a suitable medication personalized to patient diagnostics. Another study proposed that the application of monoclonal antibody, which targets the HTT protein may deplete its concentration in the cell, proving that monoclonal antibodies can interfere with the pathological processes of mHTT spreading in vivo ( 74 ).
Cell replacement therapy for HD using stem cells may be another opportunity to alleviate symptoms in patients ( 75 ). Furthermore, some case studies have indicated that exercise and physical activity may be beneficial for patients in terms of motor function, gait speed and balance, and social benefits have been also identified ( 76 ). Thus, exercise may play a complementary role in the treatment of the disorder.
5. Conclusions and future perspectives
HD is the first trinucleotide disease that was described and the first autosomal-dominant disease with a possible diagnosis prior to the manifestation of symptoms. Since 1983 and the localization of the gene, knowledge of the disorder has markedly increased, which is necessary in order to improve the quality of life of patients and improve therapeutic strategies by discovering novel molecular targets. The pathophysiology of HD is significant for designing and developing proper treatments ( 77 ). Science offers possibilities for attenuating the symptoms of the disease, and even the onset; however, it is also critical to identify effective biomarkers that may help prevent HD manifestation by early detection and blocking its course. Modern therapeutic trial design also vastly relies on identifying and examining biomarkers relevant to each disease.
The next step may be to evaluate and use data from genome-wide association studies and account for their clinical utility. Studies (as aforementioned) towards this direction have contributed to the existing knowledge concerning the association of genetic variations to the onset of symptoms and the progression of HD. The combination of early testing in order to predict the possible HD onset and new targeted and personalized medicine represents the future in preventing and hopefully, eliminating neurodegenerative diseases. The path for science ahead to help patients with HD is a long one. Until then, finding the optimal care for patients and caregivers is significant.
Acknowledgements
Funding statement.
Funding: The authors would like to acknowledge funding from the following organizations: i) AdjustEBOVGP-Dx (RIA2018EF-2081): Biochemical Adjustments of native EBOV Glycoprotein in Patient Sample to Unmask target Epitopes for Rapid Diagnostic Testing. A European and Developing Countries Clinical Trials Partnership (EDCTP2) under the Horizon 2020 ‘Research and Innovation Actions’ DESCA; ii) ‘MilkSafe: A novel pipeline to enrich formula milk using omics technologies’, a research co-financed by the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH-CREATE-INNOVATE (project code: T2EDK-02222); iii) ‘INSPIRED-The National Research Infrastructures on Integrated Structural Biology, Drug Screening Efforts and Drug Target Functional Characterization’ (Grant MIS 5002550) implemented under the Action ‘Reinforcement of the Research and Innovation Infrastructure’, funded by the Operational Program ‘Competitiveness, Entrepreneurship and Innovation’ (NSRF 2014-2020) and co-financed by Greece and the European Union (European Regional Development Fund), and iv) ‘OPENSCREENGR An Open-Access Research Infrastructure of Chemical Biology and Target-Based Screening Technologies for Human and Animal Health, Agriculture and the Environment’ (Grant MIS 5002691), implemented under the Action ‘Reinforcement of the Research and Innovation Infrastructure’, funded by the Operational Program ‘Competitiveness, Entrepreneurship and Innovation’ (NSRF 2014-2020) and co-financed by Greece and the European Union (European Regional Development Fund).
Availability of data and materials
Authors' contributions.
All authors (AMP, EP, RG, MS, TM, LP, ID, KP, KD, DAS, FB, GPC, EE and DV) contributed to the conceptualization, design, writing, drafting, revising, editing and reviewing of the manuscript. All authors have read and approved the final manuscript. Data authentication is not applicable.
Ethics approval and consent to participate
Patient consent for publication, competing interests.
DAS is the Editor-in-Chief for the journal, but had no personal involvement in the reviewing process, or any influence in terms of adjudicating on the final decision, for this article. The other authors declare that they have no competing interests.

Cognitive symptoms of Huntington’s can be tough to process
Psychosis causes a person to lose a degree of contact with reality. According to the National Health Service, three main symptoms are linked to a psychotic episode: hallucinations, delusions, and confused and disturbed thoughts. As a 2019 Huntington’s Disease News article noted, “Psychosis is defined as experiencing hallucinations…

Specific nerve cells lost early in Huntington’s disease course: Study
A type of brain nerve cells called layer 5a pyramidal neurons — especially those projecting into the striatum, a brain region greatly affected in Huntington’s disease — are lost early in the course of Huntington’s, a study found. Excessive CAG repeats in the HTT gene, the cause of…

For a patient or caregiver, understanding HIPAA is a hip thing
From time to time, my son-in-law, René, will ask me “when you were young” questions, usually to poke good-natured fun at me. Here’s what I mean: I was born in 1964, but if you heard what he’s asked me — such as, “Did you use an abacus instead of…

Featured Column How Huntington’s disease symptoms can affect employment
Columnist Becky Field shares how some of her family members lost their jobs due to the progression of Huntington’s disease symptoms.
Click the link below to find all of the latest news related to Huntington’s disease.
Perspectives

Is there a link between Huntington’s disease and liver function?
The struggles that come with a patient’s lack of mental capacity, a lazy saturday gave us a chance to enjoy life’s simple pleasures, how huntington’s disease symptoms can affect employment, newly diagnosed start your journey here..

Need to know
Becoming educated and informed about Huntington’s disease is a good first step. Here you’ll find general information about the disease, including diagnosis, symptoms, and causes.

Personal experiences
It helps to know others have been where you are now. Here’s a collection of our columnists’ words of wisdom to help you along on your Huntington’s disease journey.

There is no cure for Huntington’s disease yet, but here you can find more information about current therapeutic approaches, as well as experimental treatments that are in the pipeline.
Subscribe to our newsletter
Get regular updates to your inbox.
On the hunt for a treatment

“Huntington’s is a ticking time bomb. If one of your parents had it, you have a 50 per cent chance of inheriting the mutated gene for it. And if you have the mutated gene, then you will develop the disease,” emphasizes Dr. Massimo Pandolfo , a neurologist and co-director of The Neuro’s Clinical Research Unit, speaking about Huntington’s Disease (HD), a progressive severe, disabling neurological disorder brought on by a gene mutation. It strikes in the prime of life with a crushing decline that leads to physical, psychological, and cognitive losses.
Damaged gene
The disease is due to a genetic mutation in the HTT gene that makes a protein called huntingtin. We all have so-called CAG repeats (Cytosine, Adenine, Guanine) in this gene, but people with the disease have a greater number of CAG repeats than usual. These drive cells to manufacture a mutant form of huntingtin, which eventually causes nerve cells (neurons) to gradually break down and die in an area of the brain responsible for control of movement, cognition and emotion.
Huntington’s Disease is very difficult for affected individuals and their family. “Often, the first changes are psychiatric. Individuals behave uncharacteristically – make bad choices, change personality. Next, they develop difficulties with planning and complex tasks, progressively leading to dementia and they also develop a movement disorder with uncontrolled twists and twitches,” explains Dr. Pandolfo.
The disease may appear at different ages because of the unstable nature of the mutation. The number of CAG repeats in the HTT gene is a major factor determining when symptoms appear; the longer the repeat, the earlier the onset. As the repeat may change in size when transmitted form the affected parent, a child may develop the disease at a younger age than their parent, a phenomenon called anticipation. When starting very early, Huntington disease may result in generalized rigidity and slowness instead of the typical twists and twitches.
A common rare disease
According to the Huntington’s Society of Canada, 1 in 7,000 people in Canada are affected. Symptoms usually begin in the prime of life between 35 and 55, and life expectancy is 10 to 25 years after the start of symptoms. There are currently no drugs to slow or stop its progression.
For the first time, the Clinical Research Unit at The Neuro will conduct several clinical trials into Huntington’s Disease, continuing to expand the list of conditions covered in its clinical trials. In the past year alone, the CRU added trials for rare diseases like MOGAD and Facioscapulohumeral muscular dystrophy (FSHD) as well as a rapidly progressing prion disease known as Creutzfeldt-Jakob.
Decreasing the gene expression
“Currently, healthcare teams use medication to treat the depression, the psychiatric issues, and decrease the involuntary movements,” says Dr. Pandolfo. “But ideally we would correct the genetic defect that causes this debilitating disease.”
Some of the new approaches being investigated try to decrease the expression of the mutated gene so that it makes less of the toxic Huntingtin protein in the hopes that this will slow the progression of the disease.
“A number of studies have identified symptoms that precede obvious clinical onset of Huntington’s, that is to say the moment when individuals begin to have very subtle changes in mood and movement. Developing an effective therapy would allow us to eventually intervene at this pre-symptomatic stage and delay the onset of disease as long as possible,” says Dr. Pandolfo.
For more about studies on Huntington’s Disease at The Neuro’s Clinical Research Unit, visit cru.mcgill.ca/mvtdisorders , contact neurocog-cru.neuro@mcgill.ca or 514-398-5500 for more information.
To encourage thoughtful and respectful conversations, comments appear with first and last names (no pseudonyms) and may be published in whole or in part, at the discretion of the Reporter. Please be constructive and respectful; all comments are moderated according to the Reporter’s guidelines . We reserve the right to close comments on individual stories. Please note that the University does not endorse the opinions expressed in comments.
You might also enjoy...
Repressing a faulty gene, driving new research in rare disease, a genetic target for young-onset dementia, tracking friedreich’s ataxia.
- Type 2 Diabetes
- Heart Disease
- Digestive Health
- Multiple Sclerosis
- COVID-19 Vaccines
- Occupational Therapy
- Healthy Aging
- Health Insurance
- Public Health
- Patient Rights
- Caregivers & Loved Ones
- End of Life Concerns
- Health News
- Thyroid Test Analyzer
- Doctor Discussion Guides
- Hemoglobin A1c Test Analyzer
- Lipid Test Analyzer
- Complete Blood Count (CBC) Analyzer
- What to Buy
- Editorial Process
- Meet Our Medical Expert Board
Huntington's vs. ALS: What Are the Differences?
Frequently asked questions.
Huntington's disease (HD) and amyotrophic lateral sclerosis (ALS) are progressive neurodegenerative (nerve-dying) diseases that affect movement. Apart from this broad similarity, HD and ALS are separate conditions.
Huntington's disease is an inherited condition associated with uncontrolled movements, psychiatric disturbances, and cognitive decline. ALS, also known as Lou Gehrig's disease, is associated with muscle weakness and (eventually) complete paralysis. The vast majority of ALS cases are not inherited.
This article reviews the key differences between Huntington's disease and ALS, including their unique causes, symptoms, diagnosis, and treatment.
gorodenkoff / Getty Images
The symptoms of Huntington's disease and ALS worsen over time, although they differ in type and how fast they progress.
Huntington's Disease Symptoms
The symptoms of Huntington's disease typically become noticeable between ages 30 and 50. The condition lasts around 15 to 20 years, from the start of symptoms to full-time care dependency and death.
Symptoms are motor (movement-related), cognitive, and psychiatric. They commonly include:
- Chorea (uncontrollable movements in the face, trunk, arms, or legs)
- Impaired coordination
- Emotional disturbances, including irritability, depression, anxiety, anger, and apathy (lack of interest)
- Hallucinations (seeing or hearing things that are not there) and delusions (false beliefs)
- Cognitive decline (e.g., reduced ability to multitask)
- Weight loss from difficulties swallowing and eating
ALS Symptoms
ALS is most likely to strike between the ages of 55 and 75. The disease's hallmark symptom is muscle weakness, leading to complete paralysis and death within a matter of years.
The weakness of ALS classically begins in an arm or leg before spreading throughout the entire body. Over time, people lose their ability to dress, bathe, walk, eat, and breathe independently.
Besides physical impairment, cognitive and behavioral dysfunction, like difficulties with problem-solving, apathy, and depression, may arise in ALS, especially as the disease progresses.
A critical difference between Huntington's disease and ALS is its cause. Huntington's disease is caused by a genetic abnormality, whereas the cause of ALS is largely unknown.
What Causes Huntington's Disease?
Huntington's disease is caused by a specific genetic mutation (change in DNA sequence) in HTT, a gene that codes for the protein huntingtin . The abnormal protein that results from the altered gene causes nerve cells within certain areas of the brain (e.g., basal ganglia) to die.
Huntington's disease is an autosomal dominant disorder . If either parent has Huntington's disease, their child has a 50% chance of inheriting the mutated gene and developing it.
What Causes ALS?
ALS involves the death of motor neurons (nerve cells that control muscle movement) within a person's brain and spinal cord.
Unlike Huntington's, ALS does not usually run in families, although there is a genetic component. Over 50 genes are identified as being linked to ALS.
Interestingly, many of these genes are involved in RNA metabolism. RNA is a molecule within living cells that creates proteins using cells' genetic information. As such, scientists believe that mechanisms like RNA misprocessing and the proteins produced not folding correctly may be involved in the nerve cell death seen in ALS.
Besides genetics, various environmental factors may increase a person's chances of developing ALS. Some of these factors are:
- Exposure to lead, electromagnetic fields, agricultural chemicals, or heavy metals
- Head trauma
- Intense physical activity
The diagnosis of Huntington's disease can be confirmed with a single genetic blood test. For ALS, the diagnosis requires a bit more detective work.
Huntington's Disease Diagnosis
A person's symptoms and family history may provide the first clues to Huntington's disease. A genetic blood test that checks for the presence of the HTT mutation can then confirm the diagnosis.
The HTT gene mutation is found on chromosome 4 and is associated with increased CAG repeats.
What Is CAG?
"CAG" stands for the nucleic acids cytosine, adenine, and guanine. These are molecules in DNA that code for proteins.
Huntington's disease develops when increased CAG repeats are present. Usually, people have less than 26 CAG repeats. Those with 36 to 39 CAG repeats may or may not manifest the disease, whereas those with 40 or more repeats definitely will.
ALS Diagnosis
A diagnosis of ALS is achieved by assessing a person's symptoms and performing a neurological exam .
Various tests will be ordered to help confirm the diagnosis. Some of these diagnostic tests include:
- Magnetic resonance imaging (MRI) is a noninvasive imaging test that uses strong magnets and radio waves to create detailed pictures of the body.
- Electromyography (EMG) and nerve conduction studies measure electrical activity in your muscles and nerve cells while they're active and at rest.
- Blood tests help rule out mimicking conditions, such as thyroid disease , vitamin B12 deficiency , human immunodeficiency virus (HIV) , and some forms of cancer.
Both Huntington's disease and ALS are fatal, incurable diseases. Treatments generally focus on managing specific symptoms, preventing complications, and maintaining comfort and quality of life.
Huntington's Disease Treatment
There are several treatments , especially prescription medications, that can help individuals living with Huntington's disease manage their symptoms.
Examples of such medications include:
- Xenazine (tetrabenazine) and Austedo (deutetrabenazine) are approved for treating Huntington-associated chorea.
- Antipsychotic drugs, such as Risperdal (risperidone), help control hallucinations and delusions.
- Antidepressants , such as a selective serotonin reuptake inhibitor (SSRI) or a tricyclic antidepressant, help treat depression, which is very common in Huntington's disease.
In addition to medication, rehabilitation therapies, including exercise therapy, speech therapy , and swallow therapy , are often integral components of treatment plans.
Counseling from a psychologist about coping with the physical, cognitive, and psychiatric changes imposed by the disease is also essential, as are family and social support.
Increased Suicide Risk in Huntington's
People living with Huntington’s disease are at a higher risk of suicidal ideation (thoughts of suicide), attempting suicide, and committing suicide.
If you have suicidal thoughts, dial 988 to reach the 988 Suicide & Crisis Lifeline and connect with a trained crisis worker. Call 911 or go to your nearest emergency room if you are in immediate danger.
ALS Treatment
As with Huntington's disease, therapies for ALS are primarily supportive. They often include:
- Physical therapy helps maintain muscle strength and mobility for as long as possible.
- Speech therapy assists in communication, often with the use of special devices as the disease progresses.
- Nutritional support with a feeding tube to help stabilize body weight and prevent malnutrition.
- Breathing care , often through noninvasive ventilatory support (oxygen delivered through a tight-fitting mask) may be needed.
Four ALS drugs are approved by the Food and Drug Administration (FDA): Rilutek (riluzole), Radicava (edaravone),and Qalsody (tofersen). These drugs are intended to slow the disease down and, possibly, prolong survival.
The only way to prevent Huntington's disease in future generations is to not have children if you know you have the gene for the condition. People with a parent who has Huntington's can have a genetic test to see if they also have Huntington's. If the gene is not present, they will not transmit the condition to their children.
While there is currently no definitive way to prevent ALS, limited research suggests that eating certain foods, such as whole-grain bread, citrus fruits, and raw vegetables, may protect you against developing ALS. Clinical trials looking into other possible ALS preventive factors are also underway.
Huntington's disease and ALS are progressive, neurodegenerative diseases that differ vastly in cause, symptoms, diagnosis, and treatment.
Huntington's disease is inherited, caused by a faulty gene that can be passed down from a parent. The disease affects the body and mind, causing uncontrolled movements and psychiatric and cognitive problems. ALS primarily affects the muscles causing progressive weakness and eventual paralysis. With ALS, the cause remains unknown in most cases.
Treatments for Huntington's disease and ALS aim to maintain the person's quality of life for as long as possible. Of note, there are three FDA-approved drugs intended to help slow ALS down. No such drugs exist yet for Huntington's disease.
A Word From Verywell
If you or a loved one has been diagnosed with a neurodegenerative condition like Huntington's disease or ALS, know that it's natural to feel a whirlwind of emotions. You might exhibit anxiety, despair, anger, or even emotional numbness. These feelings can ebb and flow as you navigate the various challenges of living with such a disease.
While being diagnosed with HD or ALS is distressing, know that you are not alone. A trained therapist or psychologist, along with your healthcare provider, can help you cope with the disease's difficulties and maintain a positive and hopeful mindset.
Huntington's disease and ALS are associated with motor (movement-related) symptoms. The hallmark motor symptom of Huntington's disease is uncontrollable movements (chorea). For ALS, the hallmark symptom is widespread muscle weakness.
Huntington's disease also causes significant cognitive and psychiatric symptoms (e.g., trouble concentrating, hallucinations, depression).
There is no cure for either condition. Treatment focuses on easing symptoms and maintaining the quality of life. Currently, there are three FDA-approved ALS drugs intended to slow disease progression. There are no treatments that can help slow the progression of Huntington's disease.
Huntington's disease is an inherited condition caused by a mutated gene that leads to nerve cell death in the brain. The precise cause of ALS is unknown, although experts suspect the disease stems from a complex interaction between a person's genetic makeup and environment.
Solberg OK, Filkuková P, Frich JC, Feragen KJB. Age at death and causes of death in patients with Huntington disease in Norway in 1986-2015 . J Huntingtons Dis . 2018;7(1):77-86. doi:10.3233/JHD-170270
National Institute of Neurological Disorders and Stroke. Huntington's disease .
National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet .
Crockford C, Newton J, Lonergan K, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS . Neurology . 2018;91(15):e1370-e1380. doi:10.1212/WNL.0000000000006317
Bates GP, Dorsey R, Gusella JF, et al. Huntington disease . Nat Rev Dis Primers . 2015;1:15005. doi:10.1038/nrdp.2015.5
Fang T, Je G, Pacut P, Keyhanian K, Gao J, Ghasemi M. Gene therapy in amyotrophic lateral sclerosis . Cells . 2022;11(13):2066. doi:10.3390/cells11132066
Barmada SJ. Linking RNA dysfunction and neurodegeneration in amyotrophic lateral sclerosis . Neurotherapeutics . 2015;12(2):340-51. doi:10.1007/s13311-015-0340-3
Oskarsson B, Horton DK, Mitsumoto H. Potential environmental factors in amyotrophic lateral sclerosis . Neurol Clin. 2015;33(4):877-888. doi:10.1016/j.ncl.2015.07.009
UC Davis Health. Huntington's Disease Clinic. The Huntington gene .
Ghasemi M. Amyotrophic lateral sclerosis mimic syndromes . Iran J Neurol . 2016;15(2):85-91
National Institute of Neurological Disorders and Stroke. Huntington's disease: Hope through research . September 18, 2020
Kachian ZR, Cohen-Zimerman S, Bega D, Gordon B, Grafman J. Suicidal ideation and behavior in Huntington's disease: Systematic review and recommendations . J Affect Disord . 2019;250:319-329. doi:10.1016/j.jad.2019.03.043
Pupillo E, Bianchi E, Chiò A, et al. Amyotrophic lateral sclerosis and food intake . Amyotroph Lateral Scler Frontotemporal Degener . 2018;19(3-4):267-274. doi:10.1080/21678421.2017.1418002
By Colleen Doherty, MD Dr. Doherty is a board-certified internist and writer living with multiple sclerosis. She is based in Chicago.
New study finds potential targets at chromosome ends for degenerative disease prevention
Published online today in Science , a new study finds that telomere lengths follow a different pattern than has thus far been understood. Instead of telomere lengths falling under one general range of shortest to longest across all chromosomes, this study finds that different chromosomes have separate end-specific telomere-length distributions.
We depend on our cells being able to divide and multiply, whether it's to replace sunburnt skin or replenish our blood supply and recover from injury. Chromosomes, which carry all of our genetic instructions, must be copied in a complete way during cell division. Telomeres, which cap the ends of chromosomes, play a critical role in this cell-renewal process -- with a direct bearing on health and disease.
The enzyme telomerase plays a key role in maintaining the length of telomeres as chromosomes replicate during cell division. UC Santa Cruz professor Carol Greider has been studying telomeres and telomerase for over 30 years. The impact of the discoveries she has made over that time are why she, along with two colleagues, won the Nobel Prize in Physiology or Medicine in 2009.
So, the findings of Greider's latest study on telomeres shouldn't have surprised her. And yet, they did.
According to Greider, this discovery means we don't fully understand the molecular process that regulates telomere lengths. And that's important because of how telomere lengths affect human health: "When telomeres get to be too short, you have age-related degenerative diseases like pulmonary fibrosis, bone-marrow failure, and immunosuppression," Greider said. "On the other hand, if telomeres are too long, it predisposes you to certain types of cancer."
Kayarash Karimian, the lead author on the paper, is a former Ph.D. student in Greider's lab at the Johns Hopkins University School of Medicine. Other co-authors of this study include researchers at the Dana-Farber Cancer Institute, Harvard Medical School, and University of Pittsburgh. Greider, a distinguished professor of molecular, cell, and developmental biology at UC Santa Cruz, and a University Professor at Johns Hopkins, was the senior author on the paper and led the work.
Why length matters
Without telomerase, telomeres would get shorter and shorter as a cell divides over and over again. Over the past 30 years, research by Greider and others have confirmed that short telomeres lead to degenerative disease -- as well as shown that telomere lengths fall within a certain range.
But this paper challenges scientific consensus by showing that a singular telomere-length range is too broad. Measuring the telomeres of 147 people for this study, the researchers found in one individual that the average telomere length across all chromosomes was 4,300 bases of DNA. Then when they isolated specific chromosomes, they found most telomere lengths differed significantly from this average. In one case, lengths differed as much as 6,000 bases, which Greider describes as "jaw dropping."
Further, they found across all 147 individuals the same telomeres were most often the shortest or longest, implying telomeres on specific chromosome ends may be the first to trigger stem-cell failure.
Innovating on nanopore sequencing
To make such precise measurements at the molecular level, Greider's team used a technique invented at UC Santa Cruz called "nanopore sequencing," a revolutionary method for reading DNA and RNA that has had an immense impact on genomics research since its 2014 debut on the market as the commercial product MinION.
Nanopore technology has enabled some of the most significant advances in the genomics field, such as the completion of a gapless human genome, and sequencing of COVID-19 genomes -- making it crucial in the fight to end the pandemic. UC Santa Cruz licensed the concept for nanopore-sequencing technology to the UK-based company Oxford Nanopore Technologies, which made MinION, the first hand-held DNA sequencer.
Notably, in the eyes of nanopore sequencing's inventors, Greider's study proves that the technique's ability to advance scientific research continues to unfold. Mark Akeson, emeritus professor of biomolecular engineering at UC Santa Cruz, notes that two preprint studies that corroborate the basic findings of Greider's paper have also been posted online.
"In my opinion, this is the most important nanopore-based paper focused on human biology since the MinION was introduced," Akeson said. "It is easy to envision broad use of their telomere-length assay in the clinic."
Akeson and David Deamer, also an emeritus professor of biomolecular engineering at the Baskin School of Engineering, were honored at the Library of Congress last year for inventing nanopore sequencing. Their colleague and friend Daniel Branton, a Havard biologist and co-inventor of the technology, was honored as well.
Implications for disease prevention
Such precise DNA reads allowed Greider's team to pinpoint the sequences adjacent to telomeres and hypothesize that those areas are where telomerase is regulating length. And if that's true, Greider said those regions, and the proteins that bind there, could serve as potential targets for new drugs for preventing disease.
In addition, their process of "telomere profiling" via nanopore sequencing could serve as a model for the development of additional MinION-based assays for high-throughput drug screening.
"This accessible technique has widespread potential for use in research, diagnostics, and drug development," Greider said. "This work indicates that there are yet undiscovered mechanisms for telomere length regulation; probing these mechanisms will inform new approaches to cancer and certain degenerative diseases."
The study, "Human telomere length is chromosome end-specific and conserved across individuals," was funded by grants from the National Institutes of Health (R35CA209974 to Greider and R01HL166265), the Johns Hopkins Bloomberg Distinguished Professorship, and the National Science Foundation Graduate Research Fellowship Program.
- Human Biology
- Diseases and Conditions
- Healthy Aging
- Alzheimer's
- K-12 Education
- Huntington's Disease
- Educational Psychology
- Human genome
- Turner syndrome
- Carbohydrate
- Social psychology
- Plastic surgery
Story Source:
Materials provided by University of California - Santa Cruz . Original written by Mike Peña. Note: Content may be edited for style and length.
Journal Reference :
- Kayarash Karimian, Aljona Groot, Vienna Huso, Ramin Kahidi, Kar-Tong Tan, Samantha Sholes, Rebecca Keener, John F. McDyer, Jonathan K. Alder, Heng Li, Andreas Rechtsteiner, Carol W. Greider. Human telomere length is chromosome end–specific and conserved across individuals . Science , 2024; DOI: 10.1126/science.ado0431
Cite This Page :
Explore More
- Two Species Interbreeding Created New Butterfly
- Warming Antarctic Deep-Sea and Sea Level Rise
- Octopus Inspires New Suction Mechanism for ...
- Cities Sinking: Urban Populations at Risk
- Puzzle Solved About Ancient Galaxy
- How 3D Printers Can Give Robots a Soft Touch
- Combo of Multiple Health Stressors Harming Bees
- Methane Emission On a Cold Brown Dwarf
- Remarkable Memories of Mountain Chickadees
- Predicting Future Marine Extinctions
Trending Topics
Strange & offbeat.
IMAGES
VIDEO
COMMENTS
Huntington's disease is a hereditary neurodegenerative disorder caused by an autosomal dominant mutation. The hallmark symptom of Huntington's disease is the presence of progressive chorea ...
Keywords: chorea, antipsychotic medication, antidepressants, mood stabilizers, antisense oligonucleotides, RNAi therapies, Huntington's disease. HD is an autosomal dominant neurodegenerative disease that currently has no approved cure. HD is characterized by an increased number of CAG trinucleotide repeats in the huntingtin gene () on the ...
Read the latest research news on Huntington's Disease. Learn about genetic risks, potential new treatments and more. ... 2021 — Psychiatric and cognitive symptoms emerge at an early stage in ...
A new Nature journal article details how RNA methylation on CAG repeats is implicated in the complex mechanism underlying Huntington's disease. The article also explains how the researchers greatly reduced the progression of disease in worms and fruit flies and extended the lifespan of flies by introducing a protein into cells that removes ...
Huntington's disease is a progressive neurodegenerative disorder inherited as an autosomal-dominant trait, with onset typically occurring in mid-adult life and characterized by movement disorder ...
Devastating neurodegenerative diseases like Huntington's, Alzheimer's, and Parkinson's are all associated with protein deposits in the brain, known as amyloid. Despite extensive research ...
The journal Nature Medicine has identified a phase 3 study of pridopidine as a treatment for Huntington's disease as one of 11 clinical trials that will shape medicine in 2022 . The URMC Clinical Trials Coordination Center (CTCC) is providing global operational support for the study, which is being conducted at more than 50 sites across the U ...
CRISPR technology improves Huntington's disease symptoms in models. ScienceDaily . Retrieved April 20, 2024 from www.sciencedaily.com / releases / 2022 / 12 / 221212140550.htm
Huntington's disease (HD) has become a target of the first clinical trials for gene therapy among movement disorders with a genetic origin. More than 100 clinical trials regarding HD have been tried, but all failed, although there were some improvements limited to symptomatic support. Compared to other neurogenetic disorders, HD is known to ...
September 7, 2021. Source: Lund University. Summary: Psychiatric and cognitive symptoms emerge at an early stage in Huntington's disease. However, research so far has mainly focused on movement ...
Huntington's disease (HD) is a neurodegenerative disorder caused by an autosomal dominant mutation leading to an abnormal CAG repeat expansion. The result is the synthesis of a toxic misfolded protein, called the mutant huntingtin protein (mHTT). Most current treatments are palliative, but the latest research has expanded into multiple ...
New research led by a University of Ottawa Faculty of Medicine team is providing compelling insights into the mechanisms underlying the progression of Huntington's disease in an animal model. The ...
Abstract. The Huntington's gene on chromosome 4 has a dominantly inherited CAG trinucleotide repeat expansion, ultimately resulting in Huntington's disease (HD), a completely penetrant neurological condition. The frequency is 10-100 times higher in the population descended from Europe than in East Asia. Through various processes, including ...
13 June 2023. New research from the Stowers Institute reveals the start of Huntington's disease. Watch on. Randal Halfmann, Ph.D., discusses new research revealing the start of Huntington's disease. Stowers scientists deduce the initiating structure of the amyloid implicated in Huntington's and present a potential therapeutic treatment ...
By University of Barcelona June 21, 2023. A recent study has discovered new alterations in neural circuits, specifically the M2 cortex's different axonal projections to the superior colliculus (SC), in mouse models of Huntington's disease. This finding, alongside observed reduced functional connectivity in the brain, could provide crucial ...
Since 1999, the Huntington's Disease Society of America has committed more than $20 million to fund research, with the goal of finding effective treatments to slow Huntington's disease. Our research efforts have helped to increase the number of scientists working on HD and have shed light on many of the complex biological mechanisms involved.
First author, Professor Sarah Tabrizi (UCL Huntington's Disease Centre, UCL Queen Square Institute of Neurology, and UK Dementia Research Institute at UCL) said: "HD families have long known that ...
T he Huntington's disease (HD) community has recently experienced setbacks, but a new research report may reignite hope, from an unexpected source: the vitamin thiamine (B1), with help from ...
Huntington's disease (HD) is an inherited disorder that causes nerve cells (neurons) in parts of the brain to gradually break down and die. The disease attacks areas of the brain that help to control voluntary (intentional) movement, as well as other areas. ... A new avenue of NINDS-supported research is asking whether additional changes to ...
22 April 2024. Dr Sarah Gunn and Professor Flaviano Giorgini. A new network has been launched by the University of Leicester to drive forward research, interventions and engagement for those living with Huntington's disease. Huntington's disease is a life-limiting inherited condition that damages nerve cells in the brain causing them to stop ...
Beyond huntingtin lowering: out-of-the-box approaches for the treatment of HD. The HD pipeline is rich and varied. Let's talk about some out-of-the-box approaches for developing drugs for HD that don't involve huntingtin lowering. Kelly Andrew | November 24, 2023. Huntington's disease research news. In plain language.
The future of Huntington's disease management. Forbes and Younes suggested that as research into different treatment options progresses, there will be an opportunity to help relieve symptoms ...
2. Genetics and pathology of Huntington's disease. HD is a fatal, autosomal dominant, progressive neurodegenerative disorder characterized by severe symptoms, including motor, cognitive and psychiatric symptoms, atrophy of the basal ganglia and the cerebral cortex, and an inevitably progressive course, resulting in mortality 5-20 years following the manifestation of symptoms.
Cognitive symptoms of Huntington's can be tough to process. Psychosis causes a person to lose a degree of contact with reality. According to the National Health Service, three main symptoms are linked to a psychotic episode: hallucinations, delusions, and confused and disturbed thoughts. As a 2019 Huntington's Disease News article noted ...
And if you have the mutated gene, then you will develop the disease," emphasizes Dr. Massimo Pandolfo, a neurologist and co-director of The Neuro's Clinical Research Unit, speaking about Huntington's Disease (HD), a progressive severe, disabling neurological disorder brought on by a gene mutation. It strikes in the prime of life with a ...
The hallmark motor symptom of Huntington's disease is uncontrollable movements (chorea). For ALS, the hallmark symptom is widespread muscle weakness. Huntington's disease also causes significant cognitive and psychiatric symptoms (e.g., trouble concentrating, hallucinations, depression). Learn More The Signs and Symptoms of Huntington's Disease.
Researchers at the University of Leeds and Lancaster University in the UK have identified a new potential target for the treatment of Alzheimer's disease—PDE4B. Their work is published in ...
New study finds potential targets at chromosome ends for degenerative disease prevention. ScienceDaily . Retrieved April 15, 2024 from www.sciencedaily.com / releases / 2024 / 04 / 240411165853.htm